

Diet restriction inhibits apoptosis and HMGB1 oxidation and promotes inflammatory cell recruitment during acetaminophen hepatotoxicity
- PMID: 20811657
- PMCID: PMC2972397
- DOI: 10.2119/molmed.2010.00126
Diet restriction inhibits apoptosis and HMGB1 oxidation and promotes inflammatory cell recruitment during acetaminophen hepatotoxicity
Retraction in
-
Retraction Note: Diet restriction inhibits apoptosis and HMGB1 oxidation and promotes inflammatory cell recruitment during acetaminophen hepatotoxicity.Mol Med. 2020 Dec 30;26(1):130. doi: 10.1186/s10020-020-00262-3. Mol Med. 2020. PMID: 33380325 Free PMC article. No abstract available.
Expression of concern in
-
Expression of Concern to: Diet restriction inhibits apoptosis and HMGB1 oxidation and promotes inflammatory cell recruitment during acetaminophen hepatoxicity.Mol Med. 2020 Jan 30;26(1):13. doi: 10.1186/s10020-020-0140-z. Mol Med. 2020. PMID: 32000658 Free PMC article.
Abstract
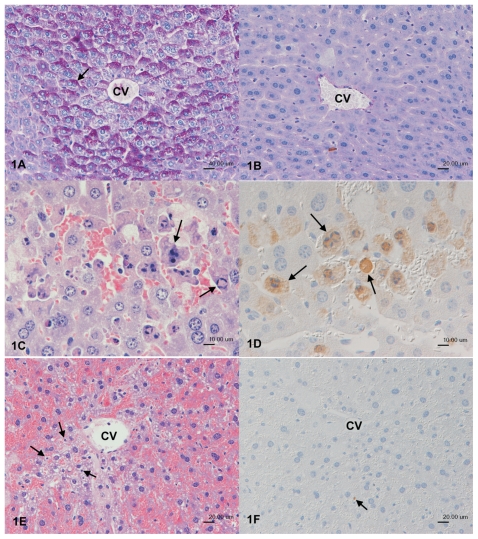
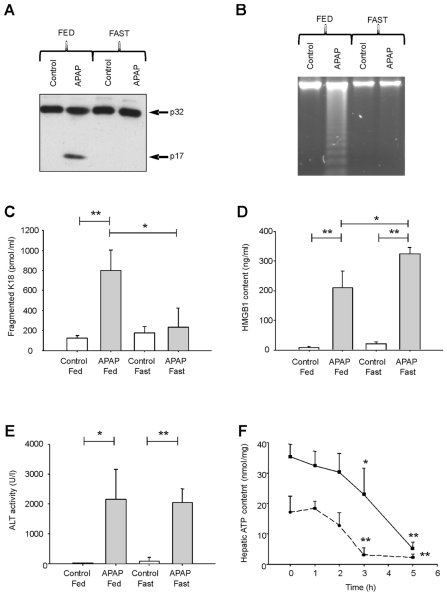
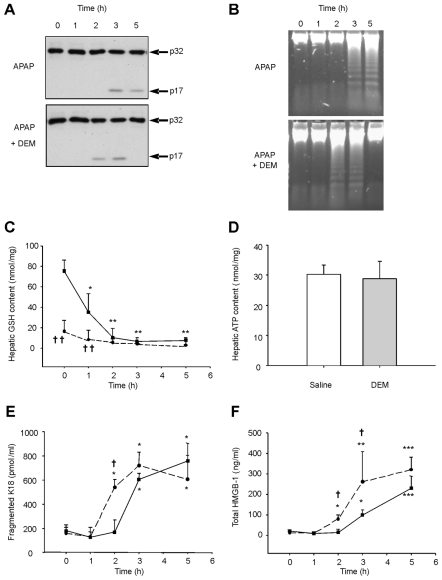
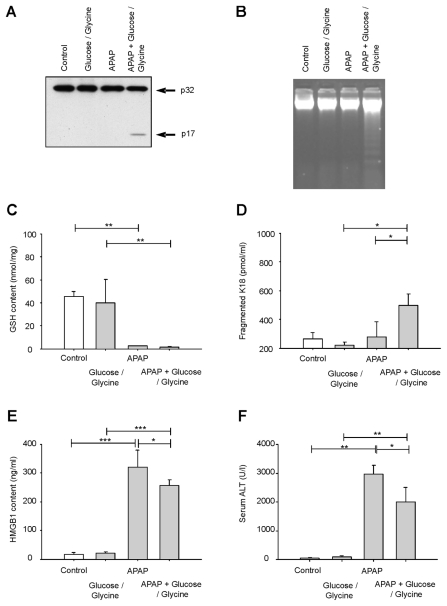
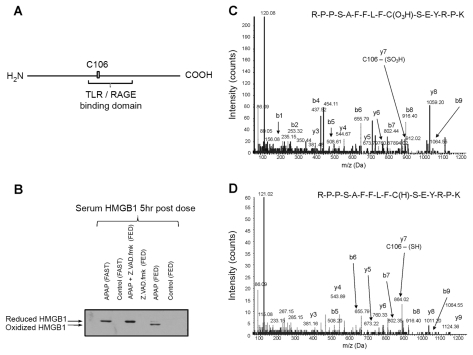
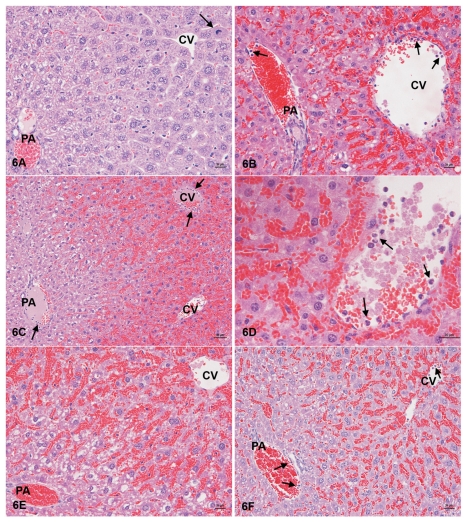
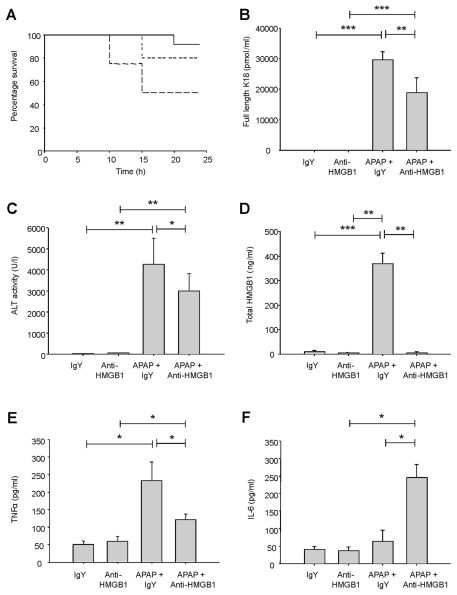
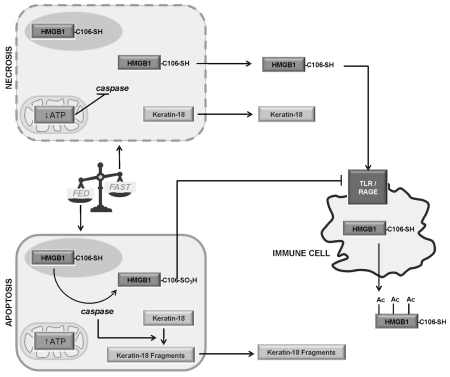
Acetaminophen (APAP) overdose is a major cause of acute liver failure and serves as a paradigm to elucidate mechanisms, predisposing factors and therapeutic interventions. The roles of apoptosis and inflammation during APAP hepatotoxicity remain controversial. We investigated whether fasting of mice for 24 h can inhibit APAP-induced caspase activation and apoptosis through the depletion of basal ATP. We also investigated in fasted mice the critical role played by inhibition of caspase-dependent cysteine 106 oxidation within high mobility group box-1 protein (HMGB1) released by ATP depletion in dying cells as a mechanism of immune activation. In fed mice treated with APAP, necrosis was the dominant form of hepatocyte death. However, apoptosis was also observed, indicated by K18 cleavage, DNA laddering and procaspase-3 processing. In fasted mice treated with APAP, only necrosis was observed. Inflammatory cell recruitment as a consequence of hepatocyte death was observed only in fasted mice treated with APAP or fed mice cotreated with a caspase inhibitor. Hepatic inflammation was also associated with loss in detection of serum oxidized-HMGB1. A significant role of HMGB1 in the induction of inflammation was confirmed with an HMGB1-neutralizing antibody. The differential response between fasted and fed mice was a consequence of a significant reduction in basal hepatic ATP, which prevented caspase processing, rather than glutathione depletion or altered APAP metabolism. Thus, the inhibition of caspase-driven apoptosis and HMGB1 oxidation by ATP depletion from fasting promotes an inflammatory response during drug-induced hepatotoxicity/liver pathology.
Figures
References
-
- Gunawan BK, Kaplowitz N. Mechanisms of drug-induced liver disease. Clin Liver Dis. 2007;11:459–75. - PubMed
-
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther. 1973;187:211–7. - PubMed
-
- Mitchell JR, et al. Acetaminophen-induced hepatic necrosis, I: role of drug metabolism. J Pharmacol Exp Ther. 1973;187:185–94. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
