

S-nitrosylation of critical protein thiols mediates protein misfolding and mitochondrial dysfunction in neurodegenerative diseases
- PMID: 20812868
- PMCID: PMC3061195
- DOI: 10.1089/ars.2010.3570
S-nitrosylation of critical protein thiols mediates protein misfolding and mitochondrial dysfunction in neurodegenerative diseases
Abstract
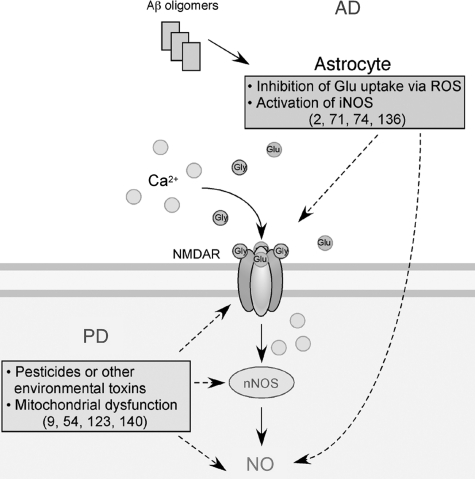
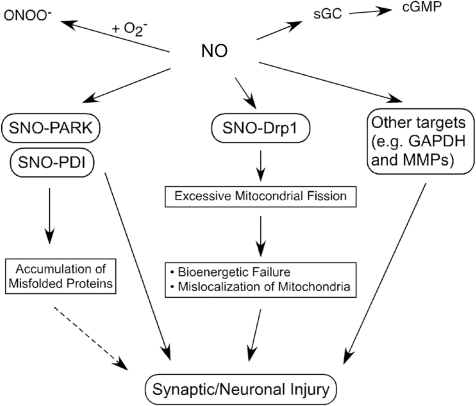
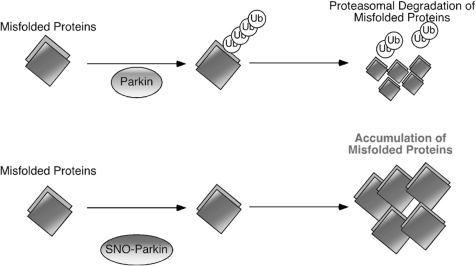
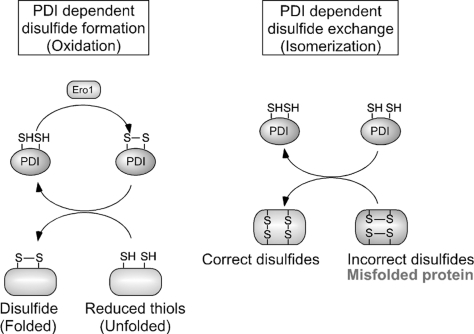
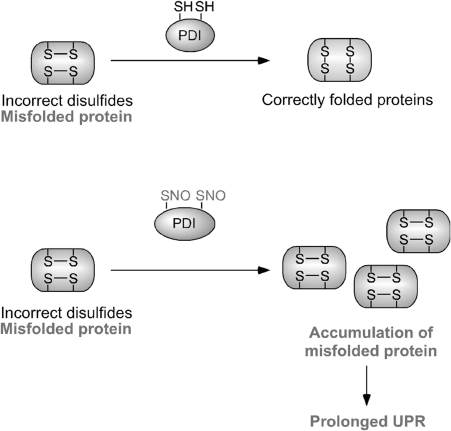
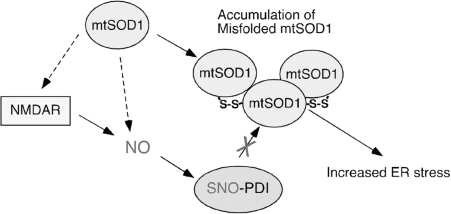
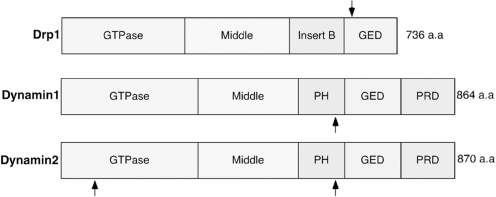
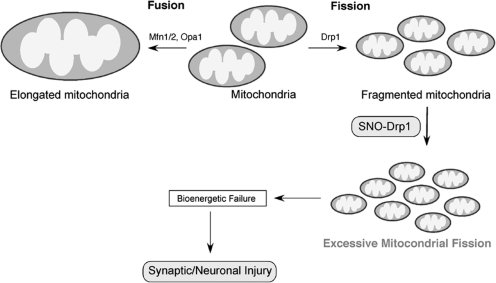
Excessive nitrosative and oxidative stress is thought to trigger cellular signaling pathways leading to neurodegenerative conditions. Such redox dysregulation can result from many cellular events, including hyperactivation of the N-methyl-D-aspartate-type glutamate receptor, mitochondrial dysfunction, and cellular aging. Recently, we and our colleagues have shown that excessive generation of free radicals and related molecules, in particular nitric oxide species (NO), can trigger pathological production of misfolded proteins, abnormal mitochondrial dynamics (comprised of mitochondrial fission and fusion events), and apoptotic pathways in neuronal cells. Emerging evidence suggests that excessive NO production can contribute to these pathological processes, specifically by S-nitrosylation of specific target proteins. Here, we highlight examples of S-nitrosylated proteins that regulate misfolded protein accumulation and mitochondrial dynamics. For instance, in models of Parkinson's disease, these S-nitrosylation targets include parkin, a ubiquitin E3 ligase and neuroprotective molecule, and protein-disulfide isomerase, a chaperone enzyme for nascent protein folding. S-Nitrosylation of protein-disulfide isomerase may also be associated with mutant Cu/Zn superoxide dismutase toxicity in amyotrophic lateral sclerosis. Additionally, in models of Alzheimer's disease, excessive NO generation leads to the formation of S-nitrosylated dynamin-related protein 1 (forming SNO-Drp1), which contributes to abnormal mitochondrial fragmentation and resultant synaptic damage.
Figures
References
-
- Akama KT. Van Eldik LJ. Beta-amyloid stimulation of inducible nitric-oxide synthase in astrocytes is interleukin-1beta- and tumor necrosis factor-alpha (TNFalpha)-dependent, and involves a TNFalpha receptor-associated factor- and NFkappaB-inducing kinase-dependent signaling mechanism. J Biol Chem. 2000;275:7918–7924. - PubMed
-
- Ankarcrona M. Dypbukt JM. Bonfoco E. Zhivotovsky B. Orrenius S. Lipton SA. Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. - PubMed
-
- Arnesano F. Banci L. Bertini I. Martinelli M. Furukawa Y. O'Halloran TV. The unusually stable quaternary structure of human Cu,Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status. J Biol Chem. 2004;279:47998–48003. - PubMed
-
- Atkin JD. Farg MA. Turner BJ. Tomas D. Lysaght JA. Nunan J. Rembach A. Nagley P. Beart PM. Cheema SS. Horne MK. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem. 2006;281:30152–30165. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
