

Generalizing moving averages for tiling arrays using combined p-value statistics
- PMID: 20812907
- PMCID: PMC2942027
- DOI: 10.2202/1544-6115.1434
Generalizing moving averages for tiling arrays using combined p-value statistics
Abstract
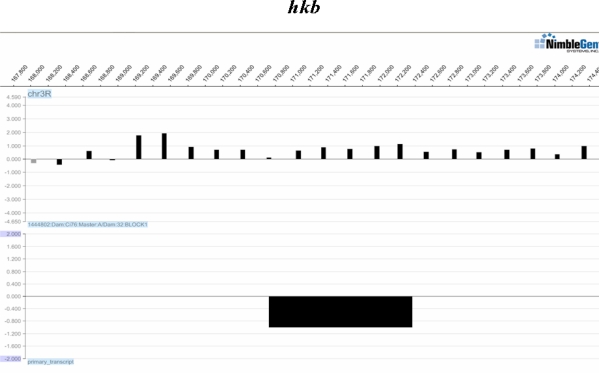
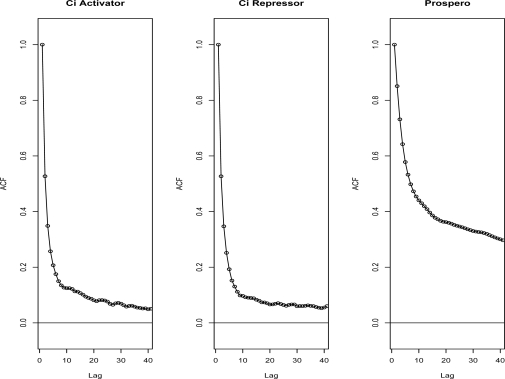
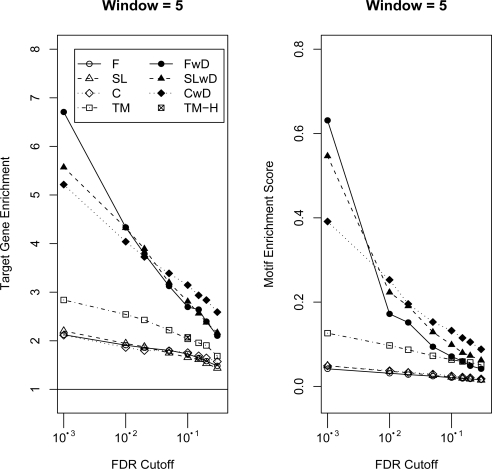
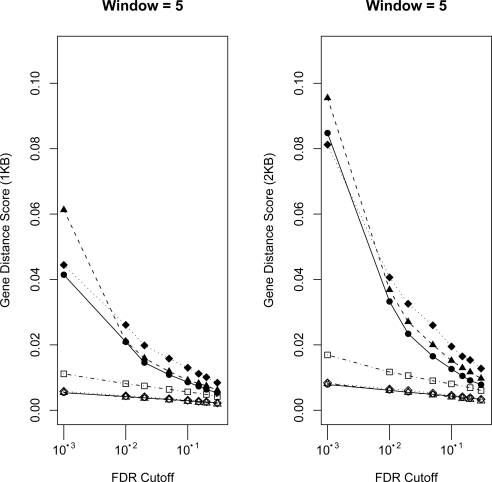
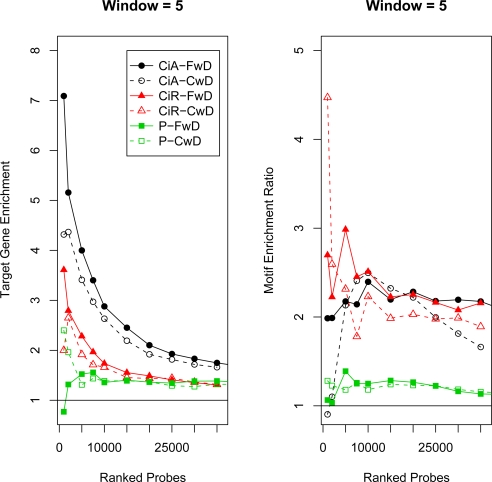
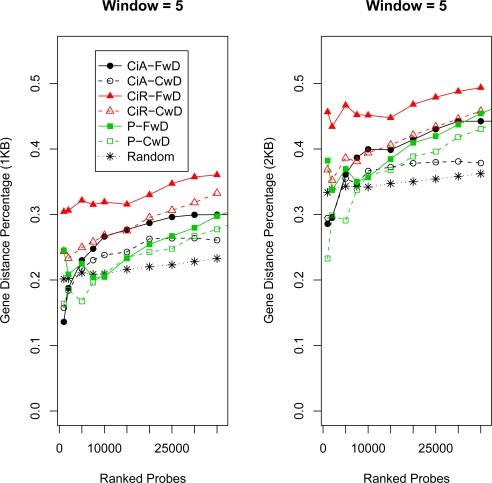
High density tiling arrays are an effective strategy for genome-wide identification of transcription factor binding regions. Sliding window methods that calculate moving averages of log ratios or t-statistics have been useful for the analysis of tiling array data. Here, we present a method that generalizes the moving average approach to evaluate sliding windows of p-values by using combined p-value statistics. In particular, the combined p-value framework can be useful in situations when taking averages of the corresponding test-statistic for the hypothesis may not be appropriate or when it is difficult to assess the significance of these averages. We exhibit the strengths of the combined p-values methods on Drosophila tiling array data and assess their ability to predict genomic regions enriched for transcription factor binding. The predictions are evaluated based on their proximity to target genes and their enrichment of known transcription factor binding sites. We also present an application for the generalization of the moving average based on integrating two different tiling array experiments.
Figures
References
-
- Alexandre C, Jacinto A, Ingham P. “Transcriptional Activation of hedgehog Target Genes in Drosophila is Mediated Directly by the Cubitus interruptus Protein, a Member of the GLI Family of Zinc Finger DNA-Binding Proteins,”. Genes and Development. 1996;10:2003–2013. doi: 10.1101/gad.10.16.2003. - DOI - PubMed
-
- Benjamini Y, Hochberg Y. “Controlling the False Discovery Rate - A Practical and Powerful Approach to Multiple Testing,”. Journal of the Royal Statistical Society Series B-Methodological. 1995;57:289–300.
-
- Bourgon R. Chromatin-Immunoprecipitation and High-Density Tiling Microarrays: A Generative Model, Methods for Analysis, and Methodology Assessment in the Absence of a ”Gold Standard”. 2006. PhD thesis, University of California, Berkeley.
-
- Brown M. “A Method for Combining Non-Independent, One-Sided Tests of Significance,”. Biometrics. 1975;31:987–992. doi: 10.2307/2529826. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases