

Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome
- PMID: 20817137
- PMCID: PMC2933349
- DOI: 10.1016/j.ajhg.2010.08.004
Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome
Abstract
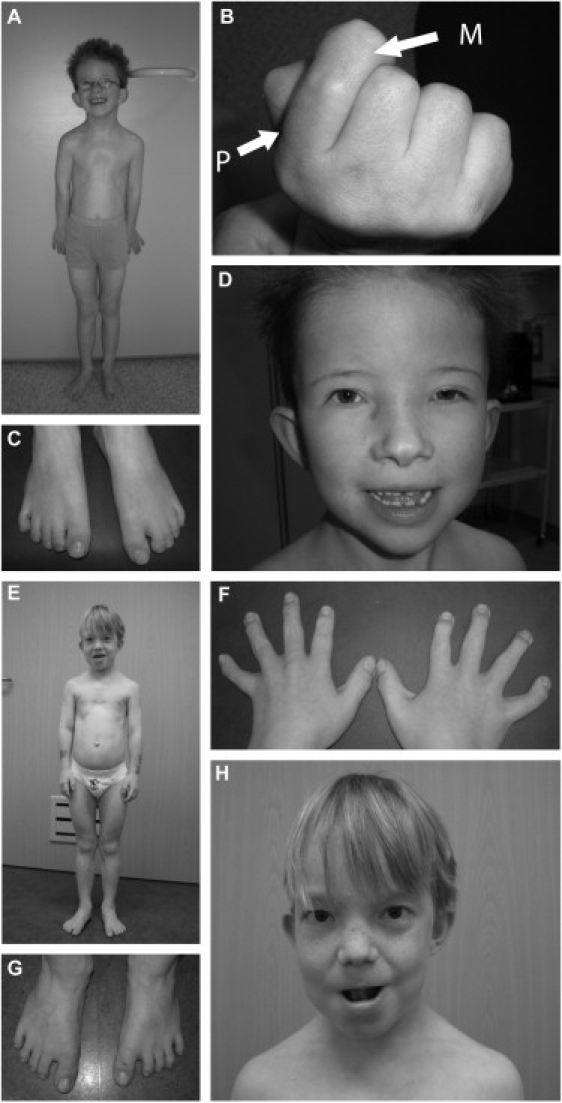
Sensenbrenner syndrome/cranioectodermal dysplasia (CED) is an autosomal-recessive disease that is characterized by craniosynostosis and ectodermal and skeletal abnormalities. We sequenced the exomes of two unrelated CED patients and identified compound heterozygous mutations in WDR35 as the cause of the disease in each of the two patients independently, showing that it is possible to find the causative gene by sequencing the exome of a single sporadic patient. With RT-PCR, we demonstrate that a splice-site mutation in exon 2 of WDR35 alters splicing of RNA on the affected allele, introducing a premature stop codon. WDR35 is homologous to TULP4 (from the Tubby superfamily) and has previously been characterized as an intraflagellar transport component, confirming that Sensenbrenner syndrome is a ciliary disorder.
2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Sensenbrenner J.A., Dorst J.P., Owens R.P. New syndrome of skeletal, dental and hair anomalies. Birth Defects Orig. Artic. Ser. 1975;11:372–379. - PubMed
-
- Levin L.S., Perrin J.C., Ose L., Dorst J.P., Miller J.D., McKusick V.A. A heritable syndrome of craniosynostosis, short thin hair, dental abnormalities, and short limbs: Cranioectodermal dysplasia. J. Pediatr. 1977;90:55–61. - PubMed
-
- Amar M.J., Sutphen R., Kousseff B.G. Expanded phenotype of cranioectodermal dysplasia (Sensenbrenner syndrome) Am. J. Med. Genet. 1997;70:349–352. - PubMed
-
- Baker K., Beales P.L. Making sense of cilia in disease: The human ciliopathies. Am. J. Med. Genet. C Semin. Med. Genet. 2009;151C:281–295. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
