

Dissecting the role of the mitochondrial chaperone mortalin in Parkinson's disease: functional impact of disease-related variants on mitochondrial homeostasis
- PMID: 20817635
- PMCID: PMC3298849
- DOI: 10.1093/hmg/ddq370
Dissecting the role of the mitochondrial chaperone mortalin in Parkinson's disease: functional impact of disease-related variants on mitochondrial homeostasis
Abstract
The mitochondrial chaperone mortalin has been linked to neurodegeneration in Parkinson's disease (PD) based on reduced protein levels in affected brain regions of PD patients and its interaction with the PD-associated protein DJ-1. Recently, two amino acid exchanges in the ATPase domain (R126W) and the substrate-binding domain (P509S) of mortalin were identified in Spanish PD patients. Here, we identified a separate and novel variant (A476T) in the substrate-binding domain of mortalin in German PD patients. To define a potential role as a susceptibility factor in PD, we characterized the functions of all three variants in different cellular models. In vitro import assays revealed normal targeting of all mortalin variants. In neuronal and non-neuronal human cell lines, the disease-associated variants caused a mitochondrial phenotype of increased reactive oxygen species and reduced mitochondrial membrane potential, which were exacerbated upon proteolytic stress. These functional impairments correspond with characteristic alterations of the mitochondrial network in cells overexpressing mutant mortalin compared with wild-type (wt), which were confirmed in fibroblasts from a carrier of the A476T variant. In line with a loss of function hypothesis, knockdown of mortalin in human cells caused impaired mitochondrial function that was rescued by wt mortalin, but not by the variants. Our genetic and functional studies of novel disease-associated variants in the mortalin gene define a loss of mortalin function, which causes impaired mitochondrial function and dynamics. Our results support the role of this mitochondrial chaperone in neurodegeneration and underscore the concept of impaired mitochondrial protein quality control in PD.
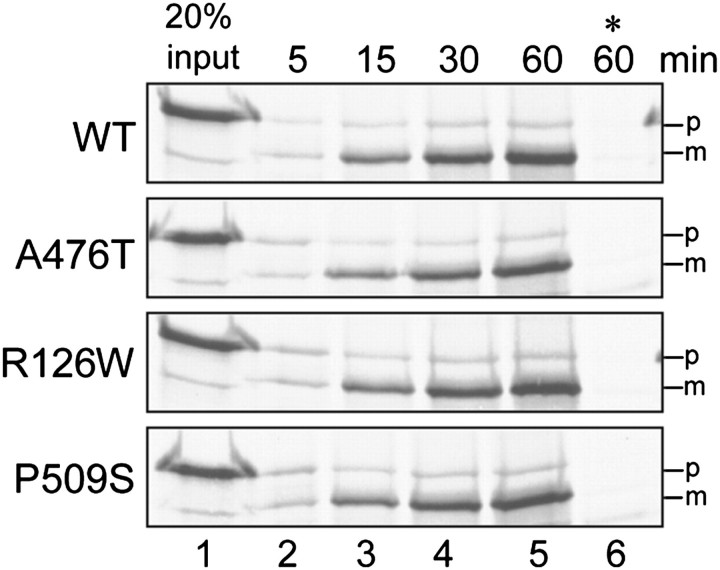
Figures







References
-
- Schiesling C., Kieper N., Seidel K., Kruger R. Review: familial Parkinson's disease—genetics, clinical phenotype and neuropathology in relation to the common sporadic form of the disease. Neuropathol. Appl. Neurobiol. 2008;34:255–271. doi:10.1111/j.1365-2990.2008.00952.x. - DOI - PubMed
-
- Simon-Sanchez J., Schulte C., Bras J.M., Sharma M., Gibbs J.R., Berg D., Paisan-Ruiz C., Lichtner P., Scholz S.W., Hernandez D.G., et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 2009;41:1308–1312. doi:10.1038/ng.487. - DOI - PMC - PubMed
-
- Schapira A.H. Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Parkinsonism Relat. Disord. 1999;5:139–143. doi:10.1016/S1353-8020(99)00029-2. - DOI - PubMed
-
- Strauss K.M., Martins L.M., Plun-Favreau H., Marx F.P., Kautzmann S., Berg D., Gasser T., Wszolek Z., Muller T., Bornemann A., et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum. Mol. Genet. 2005;14:2099–2111. doi:10.1093/hmg/ddi215. - DOI - PubMed
-
- Valente E.M., Salvi S., Ialongo T., Marongiu R., Elia A.E., Caputo V., Romito L., Albanese A., Dallapiccola B., Bentivoglio A.R. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann. Neurol. 2004;56:336–341. doi:10.1002/ana.20256. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
