

High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency
- PMID: 20818383
- PMCID: PMC2977978
- DOI: 10.1038/ng.659
High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency
Abstract
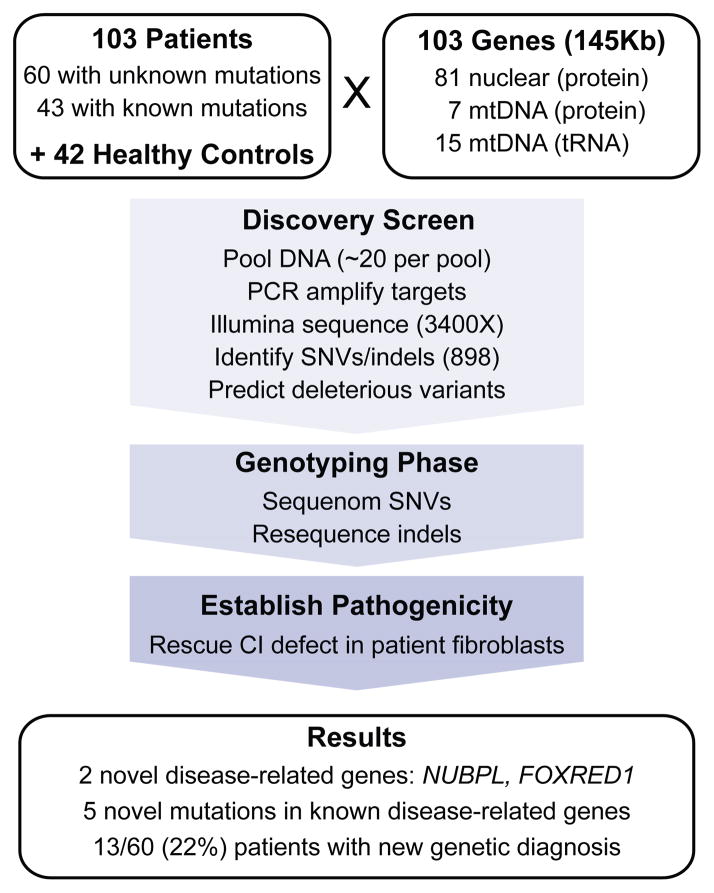
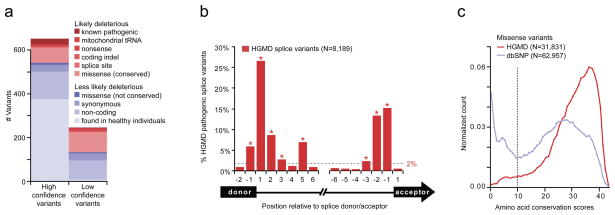
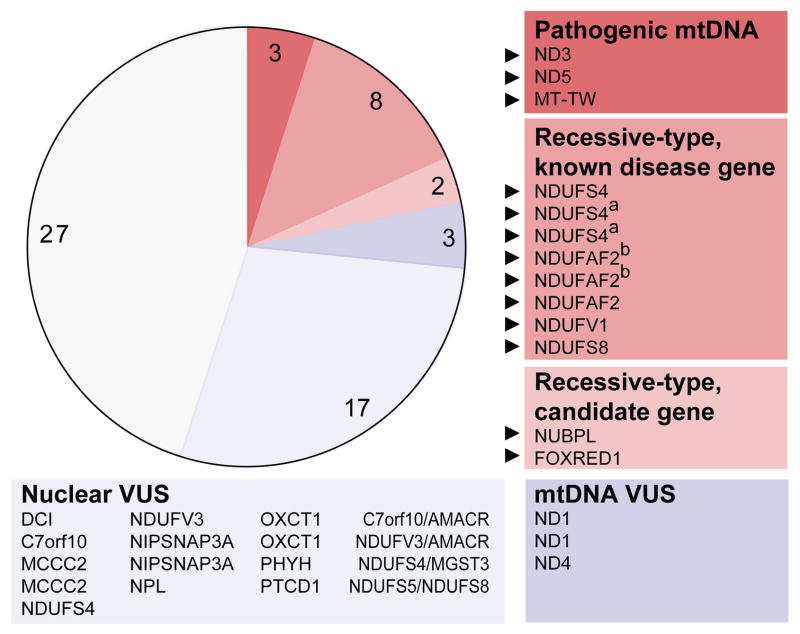
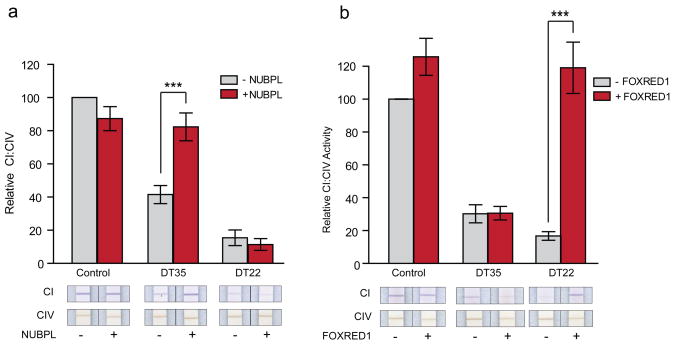
Discovering the molecular basis of mitochondrial respiratory chain disease is challenging given the large number of both mitochondrial and nuclear genes that are involved. We report a strategy of focused candidate gene prediction, high-throughput sequencing and experimental validation to uncover the molecular basis of mitochondrial complex I disorders. We created seven pools of DNA from a cohort of 103 cases and 42 healthy controls and then performed deep sequencing of 103 candidate genes to identify 151 rare variants that were predicted to affect protein function. We established genetic diagnoses in 13 of 60 previously unsolved cases using confirmatory experiments, including cDNA complementation to show that mutations in NUBPL and FOXRED1 can cause complex I deficiency. Our study illustrates how large-scale sequencing, coupled with functional prediction and experimental validation, can be used to identify causal mutations in individual cases.
Figures

References
-
- Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126:1905–12. - PubMed
-
- Distelmaier F, et al. Mitochondrial complex I deficiency: from organelle dysfunction to clinical disease. Brain. 2009;132:833–42. - PubMed
-
- Janssen RJ, Nijtmans LG, van den Heuvel LP, Smeitink JA. Mitochondrial complex I: structure, function and pathology. J Inherit Metab Dis. 2006;29:499–515. - PubMed
-
- Lazarou M, Thorburn DR, Ryan MT, McKenzie M. Assembly of mitochondrial complex I and defects in disease. Biochim Biophys Acta. 2009;1793:78–88. - PubMed
-
- Bernier FP, et al. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology. 2002;59:1406–11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
