

Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline
- PMID: 20823466
- PMCID: PMC2936060
- DOI: 10.1210/jc.2009-2631
Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline
Erratum in
- J Clin Endocrinol Metab. 2010 Nov;95(11):5137
-
Corrigendum to: "Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline".J Clin Endocrinol Metab. 2021 Jun 16;106(7):e2853. doi: 10.1210/clinem/dgab316. J Clin Endocrinol Metab. 2021. PMID: 33964163 No abstract available.
Abstract
Objective: We developed clinical practice guidelines for congenital adrenal hyperplasia (CAH).
Participants: The Task Force included a chair, selected by The Endocrine Society Clinical Guidelines Subcommittee (CGS), ten additional clinicians experienced in treating CAH, a methodologist, and a medical writer. Additional experts were also consulted. The authors received no corporate funding or remuneration.
Consensus process: Consensus was guided by systematic reviews of evidence and discussions. The guidelines were reviewed and approved sequentially by The Endocrine Society's CGS and Clinical Affairs Core Committee, members responding to a web posting, and The Endocrine Society Council. At each stage, the Task Force incorporated changes in response to written comments.
Conclusions: We recommend universal newborn screening for severe steroid 21-hydroxylase deficiency followed by confirmatory tests. We recommend that prenatal treatment of CAH continue to be regarded as experimental. The diagnosis rests on clinical and hormonal data; genotyping is reserved for equivocal cases and genetic counseling. Glucocorticoid dosage should be minimized to avoid iatrogenic Cushing's syndrome. Mineralocorticoids and, in infants, supplemental sodium are recommended in classic CAH patients. We recommend against the routine use of experimental therapies to promote growth and delay puberty; we suggest patients avoid adrenalectomy. Surgical guidelines emphasize early single-stage genital repair for severely virilized girls, performed by experienced surgeons. Clinicians should consider patients' quality of life, consulting mental health professionals as appropriate. At the transition to adulthood, we recommend monitoring for potential complications of CAH. Finally, we recommend judicious use of medication during pregnancy and in symptomatic patients with nonclassic CAH.
Figures
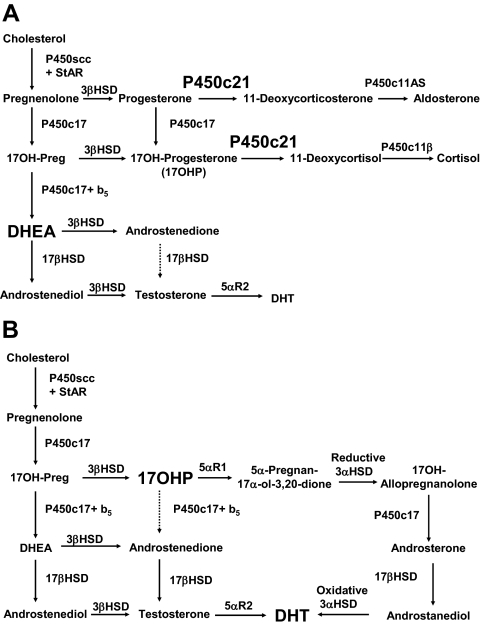
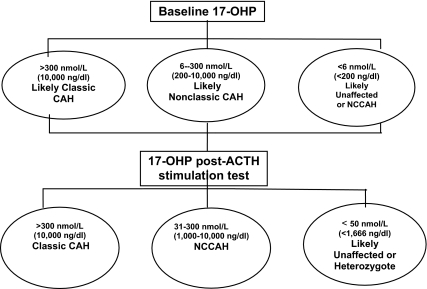
References
-
- Nordenström A, Ahmed S, Jones J, Coleman M, Price DA, Clayton PE, Hall CM 2005 Female preponderance in congenital adrenal hyperplasia due to CYP21 deficiency in England: implications for neonatal screening. Horm Res 63:22–28 - PubMed
-
- Pang S, Shook MK 1997 Current status of neonatal screening for congenital adrenal hyperplasia. Curr Opin Pediatr 9:419–423 - PubMed
-
- Therrell BL 2001 Newborn screening for congenital adrenal hyperplasia. Endocrinol Metab Clin North Am 30:15–30 - PubMed
-
- van der Kamp HJ, Wit JM 2004 Neonatal screening for congenital adrenal hyperplasia. Eur J Endocrinol 151(Suppl 3):U71–U75 - PubMed
-
- Krone N, Dhir V, Ivison HE, Arlt W 2007 Congenital adrenal hyperplasia and P450 oxidoreductase deficiency. Clin Endocrinol (Oxf) 66:162–172 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
