

Tyramide signal amplification for analysis of kinase activity by intracellular flow cytometry
- PMID: 20824632
- PMCID: PMC3036012
- DOI: 10.1002/cyto.a.20970
Tyramide signal amplification for analysis of kinase activity by intracellular flow cytometry
Abstract
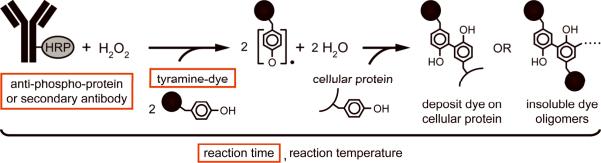
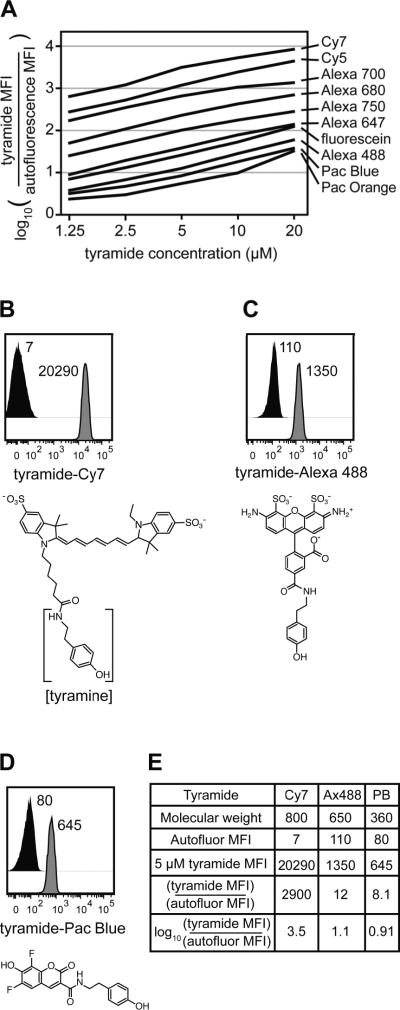
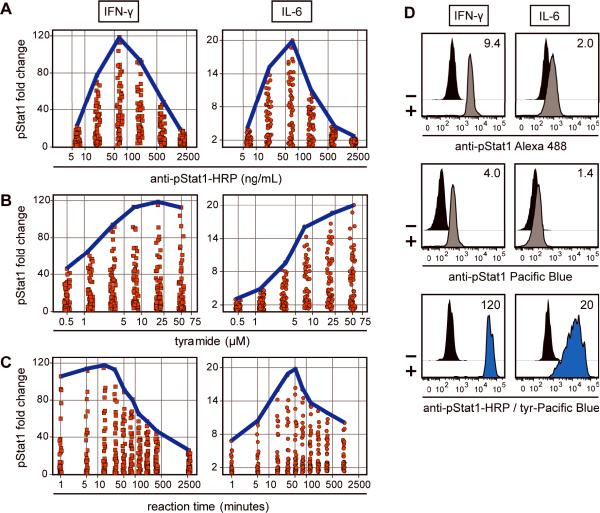
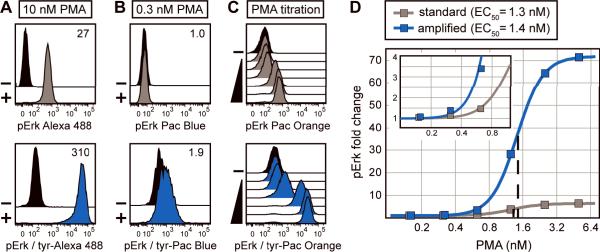
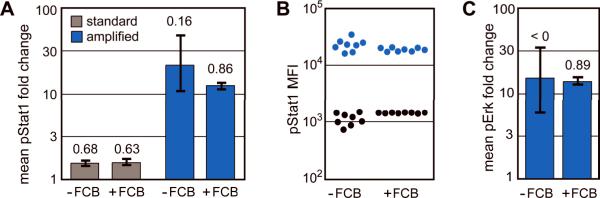
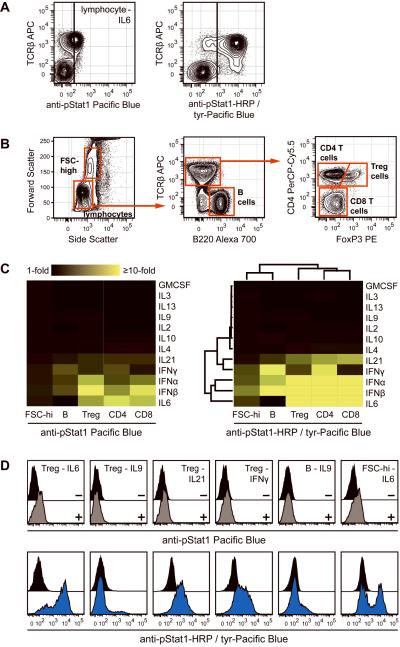
Intracellular flow cytometry permits quantitation of diverse molecular targets at the single-cell level. However, limitations in detection sensitivity inherently restrict the method, sometimes resulting in the inability to measure proteins of very low abundance or to differentiate cells expressing subtly different protein concentrations. To improve these measurements, an enzymatic amplification approach called tyramide signal amplification (TSA) was optimized for assessment of intracellular kinase cascades. First, Pacific Blue, Pacific Orange, and Alexa Fluor 488 tyramide reporters were shown to exhibit low nonspecific binding in permeabilized cells. Next, the effects of antibody concentration, tyramide concentration, and reaction time on assay resolution were characterized. Use of optimized TSA resulted in a 10-fold or greater improvement in measurement resolution of endogenous Erk and Stat cell signaling pathways relative to standard, nonamplified detection. TSA also enhanced assay sensitivity and, in conjunction with fluorescent cell barcoding, improved assay performance according to a metric used to evaluate high-throughput drug screens. TSA was used to profile Stat1 phosphorylation in primary immune system cells, which revealed heterogeneity in various populations, including CD4+ FoxP3+ regulatory T cells. We anticipate the approach will be broadly applicable to intracellular flow cytometry assays with low signal-to-noise ratios.
© 2010 International Society for Advancement of Cytometry.
Figures
References
-
- Bauer KD, Jacobberger JW. Analysis of intracellular proteins. Methods Cell Biol. 1994;41:351–76. - PubMed
-
- Krutzik PO, Nolan GP. Intracellular phospho-protein staining techniques for flow cytometry: monitoring single cell signaling events. Cytometry A. 2003;55A:61–70. - PubMed
-
- Schulz KR, Danna EA, Krutzik PO, Nolan GP. Single-cell phospho-protein analysis by flow cytometry. Curr Protoc Immunol. 2007;Chapter 8(Unit 8):17. - PubMed
-
- Lamoreaux L, Roederer M, Koup R. Intracellular cytokine optimization and standard operating procedure. Nat Protoc. 2006;1:1507–16. - PubMed
-
- Perez OD, Nolan GP. Simultaneous measurement of multiple active kinase states using polychromatic flow cytometry. Nat Biotechnol. 2002;20:155–62. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
