

AT-1 is the ER membrane acetyl-CoA transporter and is essential for cell viability
- PMID: 20826464
- PMCID: PMC2939804
- DOI: 10.1242/jcs.068841
AT-1 is the ER membrane acetyl-CoA transporter and is essential for cell viability
Abstract
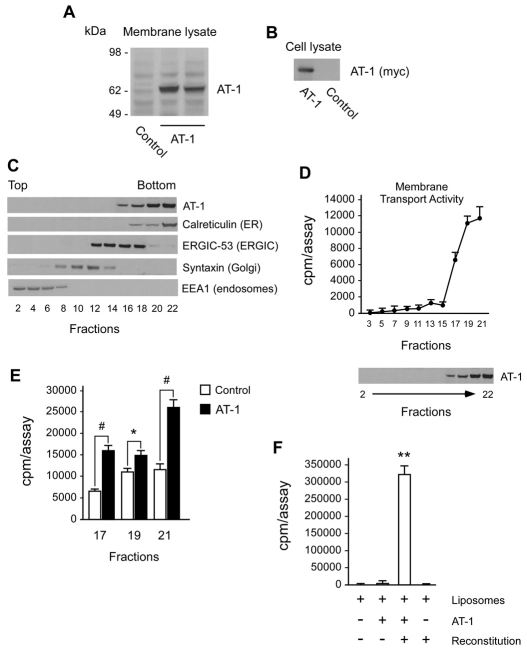
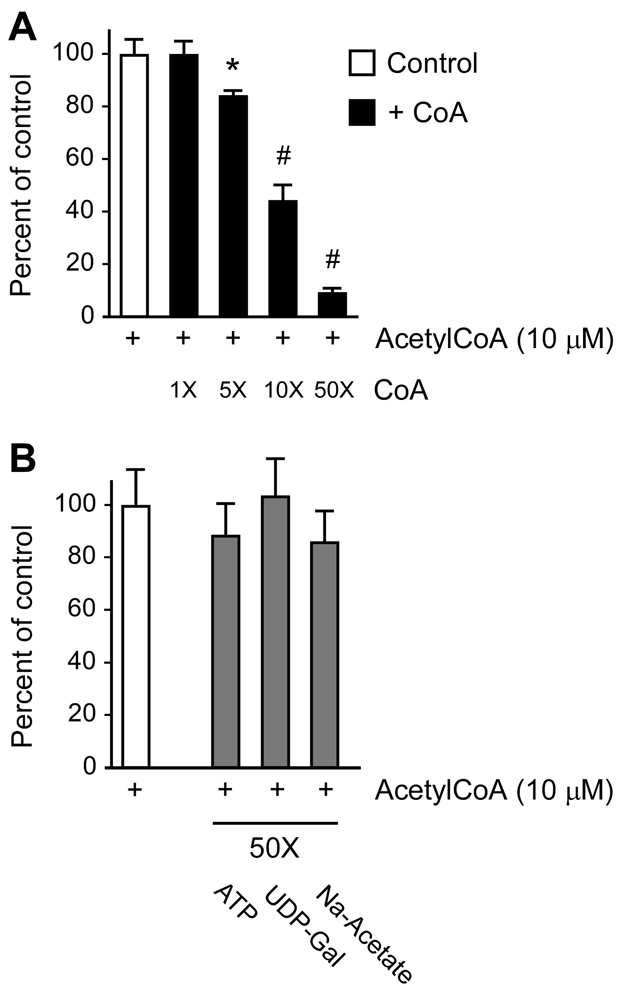
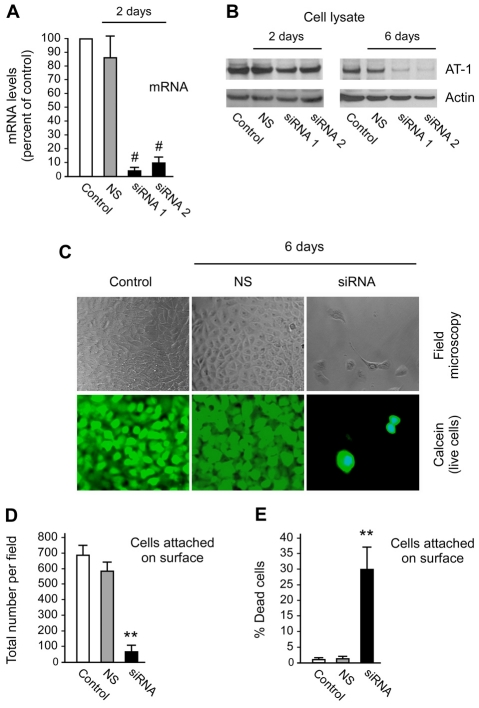
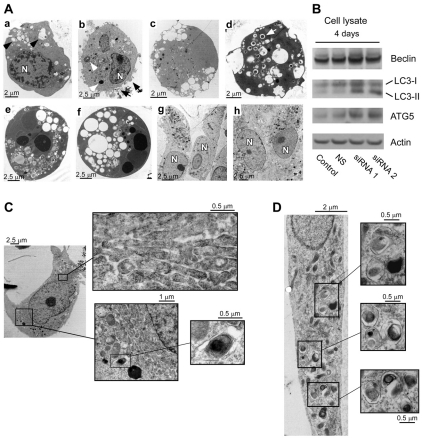
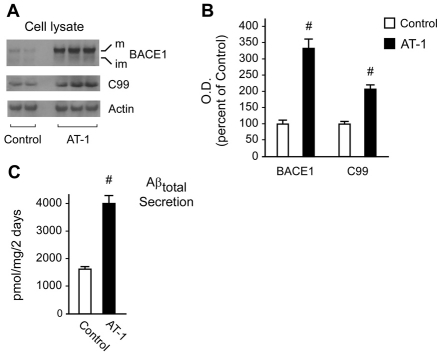
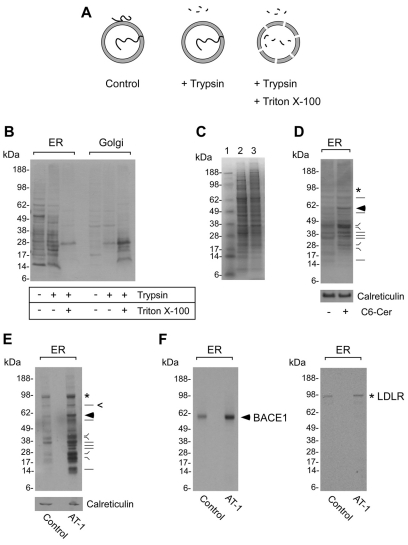
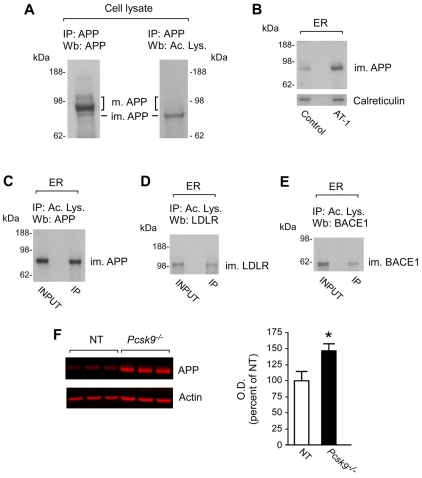
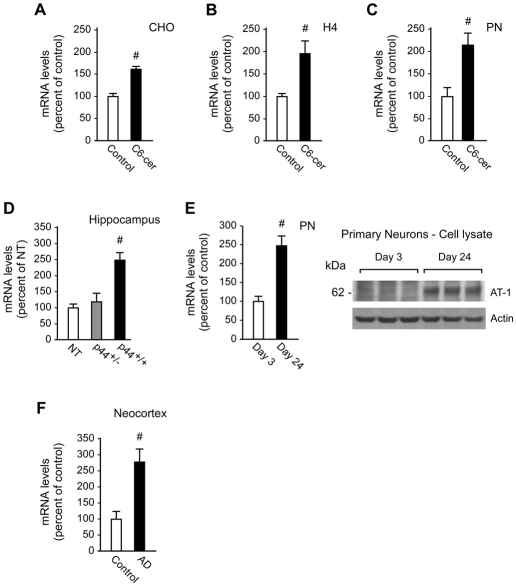
The transient or permanent modification of nascent proteins in the early secretory pathway is an essential cellular function that ensures correct folding and maturation of membrane and secreted proteins. We have recently described a new form of post-translational regulation of the membrane protein β-site APP cleaving enzyme 1 (BACE1) involving transient lysine acetylation in the lumen of the endoplasmic reticulum (ER). The essential components of this process are two ER-based acetyl-CoA:lysine acetyltransferases, ATase1 and ATase2, and a membrane transporter that translocates acetyl-CoA into the lumen of the ER. Here, we report the functional identification of acetyl-CoA transporter 1 (AT-1) as the ER membrane acetyl-CoA transporter. We show that AT-1 regulates the acetylation status of ER-transiting proteins, including the membrane proteins BACE1, low-density lipoprotein receptor and amyloid precursor protein (APP). Finally, we show that AT-1 is essential for cell viability as its downregulation results in widespread cell death and induction of features characteristic of autophagy.
Figures
References
-
- Abisambra J. F., Fiorelli T., Padmanabhan J., Wefes I., Neame P., Goodwin B., Norden M., Potter H. (2009). APP over-expression affects the localization of PCSK9 and LDLR. Program no. 43.5/D26, Society for Neuroscience 2009 Annual Meeting, Chicago, IL
-
- Axe E. L., Walker S. A., Manifava M., Chandra P., Roderick H. L., Habermann A., Griffiths G., Ktistakis N. T. (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685-701 - PMC - PubMed
-
- Berninsone P. M., Hirschberg C. B. (2000). Nucleotide sugar transporters of the Golgi apparatus. Curr. Opin. Struct. Biol. 10, 542-547 - PubMed
-
- Carey D. J., Hirschberg C. B. (1981). Topography of sialoglycoproteins and sialyltransferases in mouse and rat liver Golgi. J. Biol. Chem. 256, 989-993 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
