

Reduced survival of motor neuron (SMN) protein in motor neuronal progenitors functions cell autonomously to cause spinal muscular atrophy in model mice expressing the human centromeric (SMN2) gene
- PMID: 20826664
- PMCID: PMC2944776
- DOI: 10.1523/JNEUROSCI.2208-10.2010
Reduced survival of motor neuron (SMN) protein in motor neuronal progenitors functions cell autonomously to cause spinal muscular atrophy in model mice expressing the human centromeric (SMN2) gene
Abstract
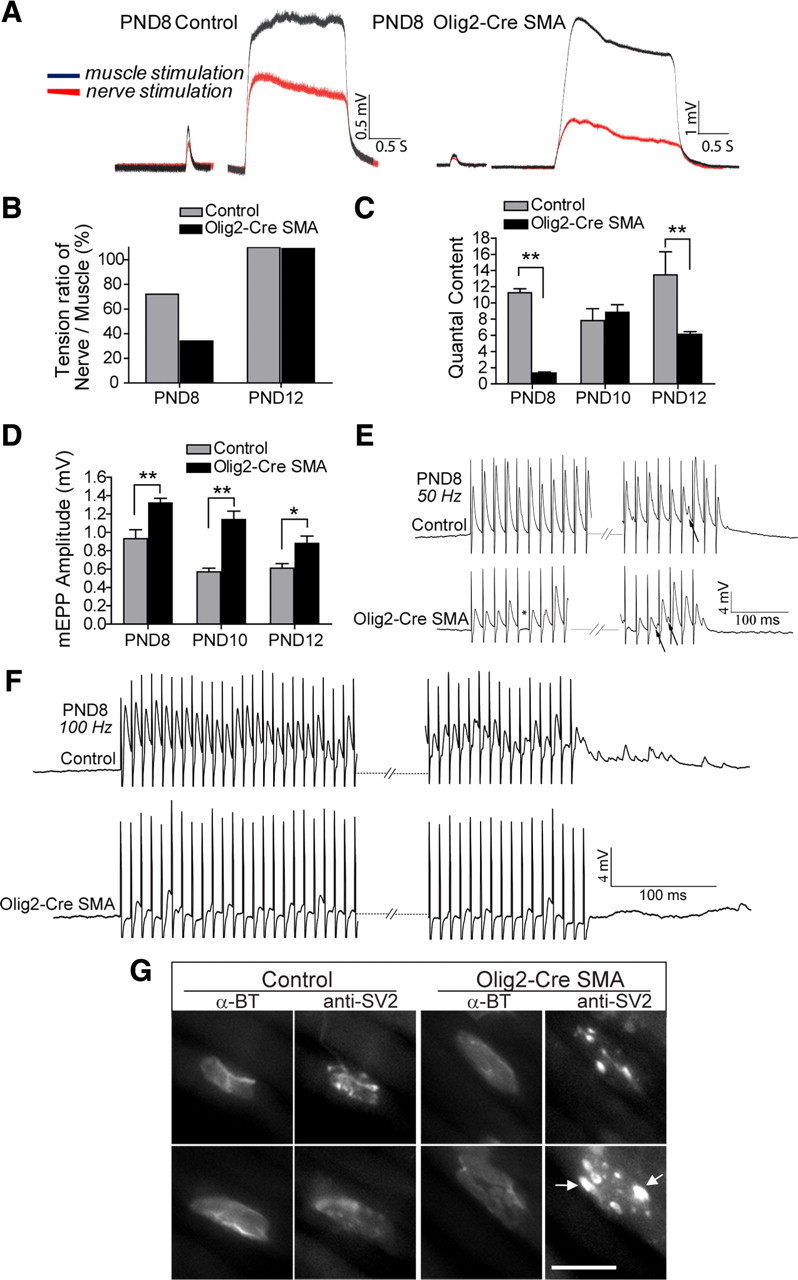
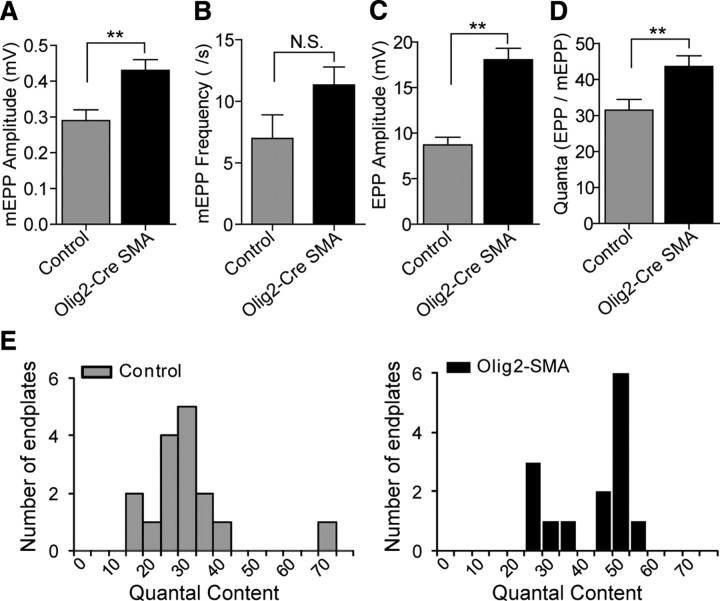
Spinal muscular atrophy (SMA) is a common (approximately 1:6400) autosomal recessive neuromuscular disorder caused by a paucity of the survival of motor neuron (SMN) protein. Although widely recognized to cause selective spinal motor neuron loss when deficient, the precise cellular site of action of the SMN protein in SMA remains unclear. In this study we sought to determine the consequences of selectively depleting SMN in the motor neurons of model mice. Depleting but not abolishing the protein in motor neuronal progenitors causes an SMA-like phenotype. Neuromuscular weakness in the model mice is accompanied by peripheral as well as central synaptic defects, electrophysiological abnormalities of the neuromuscular junctions, muscle atrophy, and motor neuron degeneration. However, the disease phenotype is more modest than that observed in mice expressing ubiquitously low levels of the SMN protein, and both symptoms as well as early electrophysiological abnormalities that are readily apparent in neonates were attenuated in an age-dependent manner. We conclude that selective knock-down of SMN in motor neurons is sufficient but may not be necessary to cause a disease phenotype and that targeting these cells will be a requirement of any effective therapeutic strategy. This realization is tempered by the relatively mild SMA phenotype in our model mice, one explanation for which is the presence of normal SMN levels in non-neuronal tissue that serves to modulate disease severity.
Figures
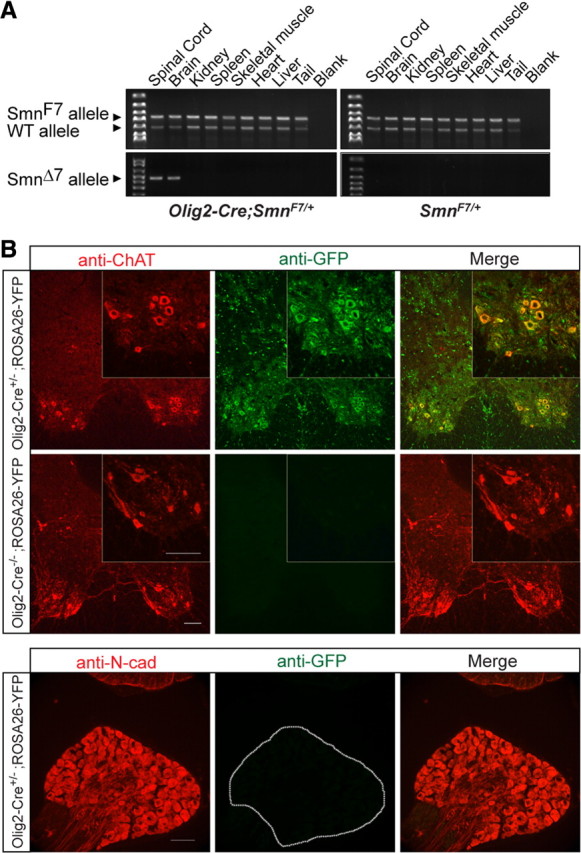
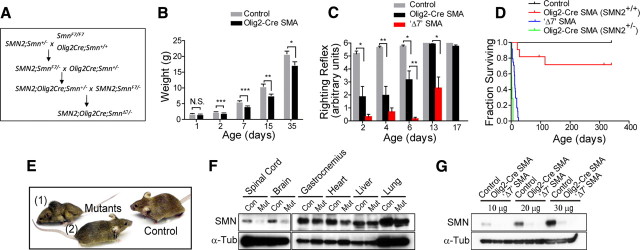
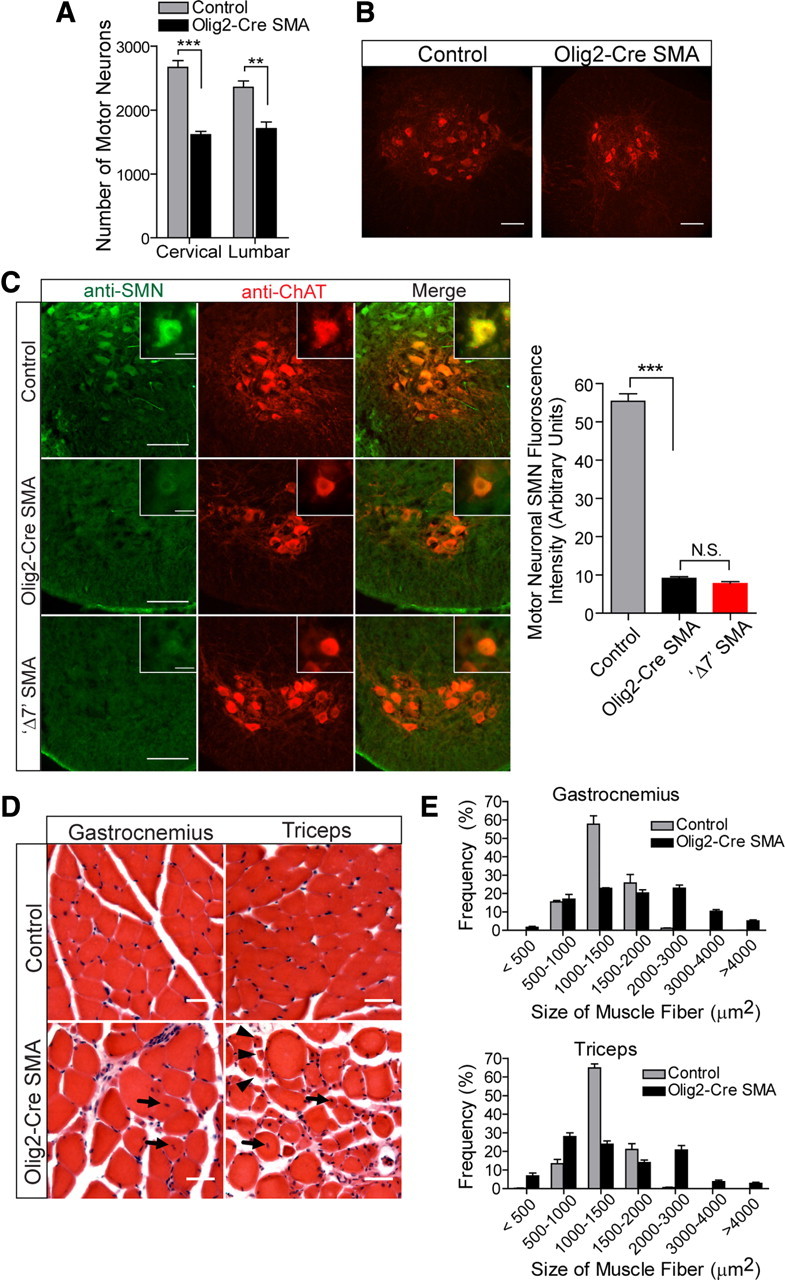
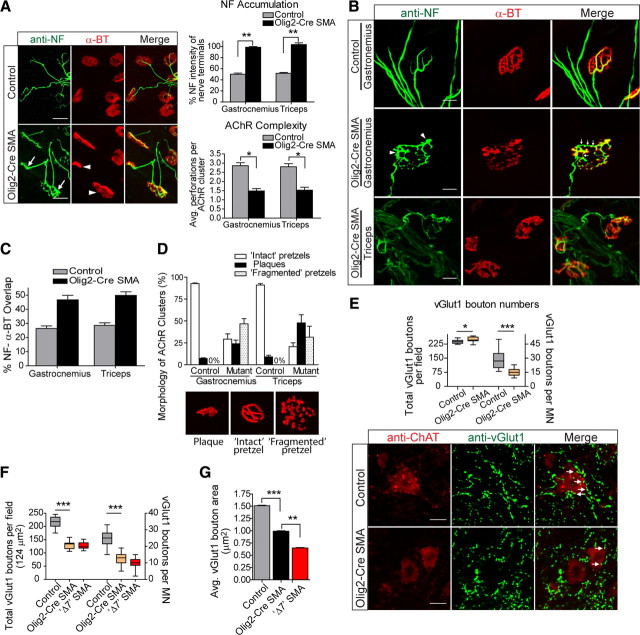
References
-
- Arber S, Ladle DR, Lin JH, Frank E, Jessell TM. ETS gene Er81 controls the formation of functional connections between group Ia sensory afferents and motor neurons. Cell. 2000;101:485–498. - PubMed
-
- Bowerman M, Anderson CL, Beauvais A, Boyl PP, Witke W, Kothary R. SMN, profilin IIa and plastin 3: a link between the deregulation of actin dynamics and SMA pathogenesis. Mol Cell Neurosci. 2009;42:66–74. - PubMed
-
- Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30:377–384. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases