

Plasma kallikrein promotes epidermal growth factor receptor transactivation and signaling in vascular smooth muscle through direct activation of protease-activated receptors
- PMID: 20826789
- PMCID: PMC2966134
- DOI: 10.1074/jbc.M110.171769
Plasma kallikrein promotes epidermal growth factor receptor transactivation and signaling in vascular smooth muscle through direct activation of protease-activated receptors
Erratum in
- J Biol Chem. 2011 Jul 1;286(26):23620. El-Shewy, Hesham M [added]
Abstract
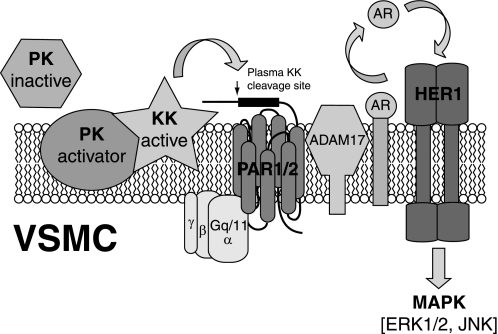
The kallikrein-kinin system, along with the interlocking renin-angiotensin system, is a key regulator of vascular contractility and injury response. The principal effectors of the kallikrein-kinin system are plasma and tissue kallikreins, proteases that cleave high molecular weight kininogen to produce bradykinin. Most of the cellular actions of kallikrein (KK) are thought to be mediated by bradykinin, which acts via G protein-coupled B1 and B2 bradykinin receptors on VSMCs and endothelial cells. Here, we find that primary aortic vascular smooth muscle but not endothelial cells possess the ability to activate plasma prekallikrein. Surprisingly, exposing VSMCs to prekallikrein leads to activation of the ERK1/2 mitogen-activated protein kinase cascade via a mechanism that requires kallikrein activity but does not involve bradykinin receptors. In transfected HEK293 cells, we find that plasma kallikrein directly activates G protein-coupled protease-activated receptors (PARs) 1 and 2, which possess consensus kallikrein cleavage sites, but not PAR4. In vascular smooth muscles, KK stimulates ADAM (a disintegrin and metalloprotease) 17 activity via a PAR1/2 receptor-dependent mechanism, leading sequentially to release of the endogenous ADAM17 substrates, amphiregulin and tumor necrosis factor-α, metalloprotease-dependent transactivation of epidermal growth factor receptors, and metalloprotease and epidermal growth factor receptor-dependent ERK1/2 activation. These results suggest a novel mechanism of bradykinin-independent kallikrein action that may contribute to the regulation of vascular responses in pathophysiologic states, such as diabetes mellitus.
Figures







References
-
- Sainz I. M., Pixley R. A., Colman R. W. (2007) Thromb. Haemost. 98, 77–83 - PubMed
-
- Hermann A., Arnhold M., Kresse H., Neth P., Fink E. (1999) Immunopharmacology 45, 135–139 - PubMed
-
- Motta G., Rojkjaer R., Hasan A. A., Cines D. B., Schmaier A. H. (1998) Blood 91, 516–528 - PubMed
-
- Motta G., Shariat-Madar Z., Mahdi F., Sampaio C. A., Schmaier A. H. (2001) Thromb. Haemost. 86, 840–847 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
