

Correlating gene expression variation with cis-regulatory polymorphism in Saccharomyces cerevisiae
- PMID: 20829281
- PMCID: PMC2953268
- DOI: 10.1093/gbe/evq054
Correlating gene expression variation with cis-regulatory polymorphism in Saccharomyces cerevisiae
Abstract
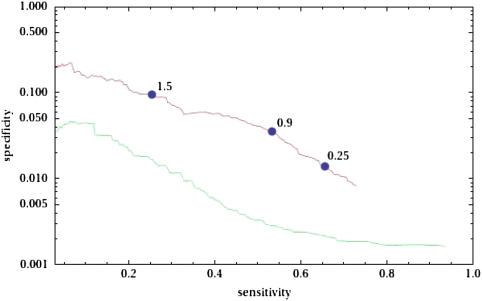
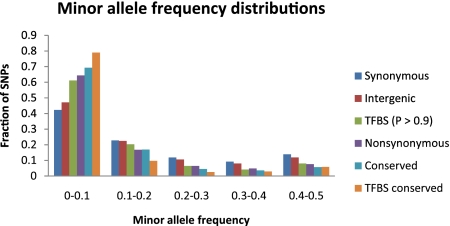
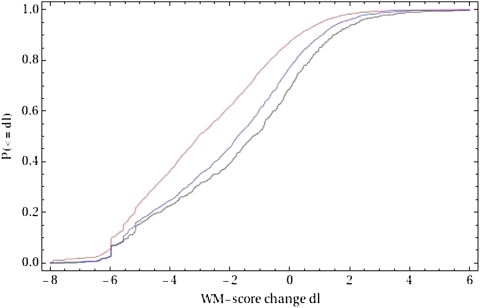
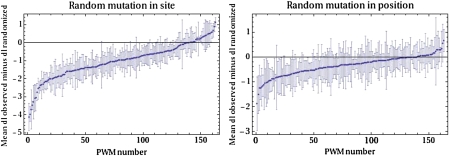
Identifying the nucleotides that cause gene expression variation is a critical step in dissecting the genetic basis of complex traits. Here, we focus on polymorphisms that are predicted to alter transcription factor binding sites (TFBSs) in the yeast, Saccharomyces cerevisiae. We assembled a confident set of transcription factor motifs using recent protein binding microarray and ChIP-chip data and used our collection of motifs to predict a comprehensive set of TFBSs across the S. cerevisiae genome. We used a population genomics analysis to show that our predictions are accurate and significantly improve on our previous annotation. Although predicting gene expression from sequence is thought to be difficult in general, we identified a subset of genes for which changes in predicted TFBSs correlate well with expression divergence between yeast strains. Our analysis thus demonstrates both the accuracy of our new TFBS predictions and the feasibility of using simple models of gene regulation to causally link differences in gene expression to variation at individual nucleotides.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
