

The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search
- PMID: 20829449
- PMCID: PMC3101864
- DOI: 10.1074/mcp.M110.003731
The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search
Abstract
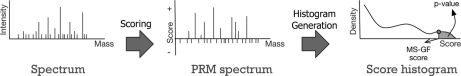
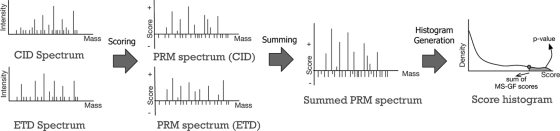
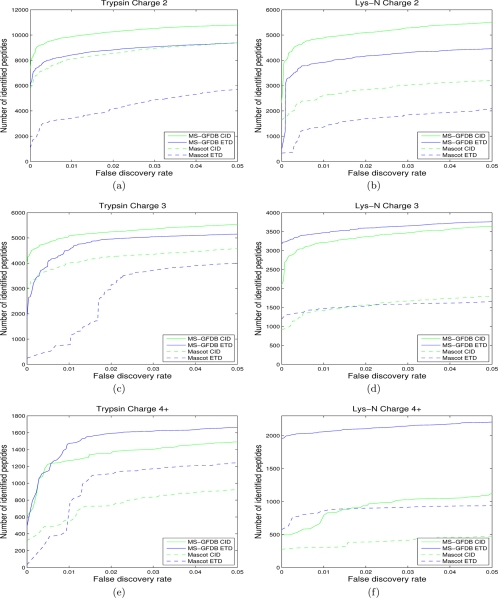
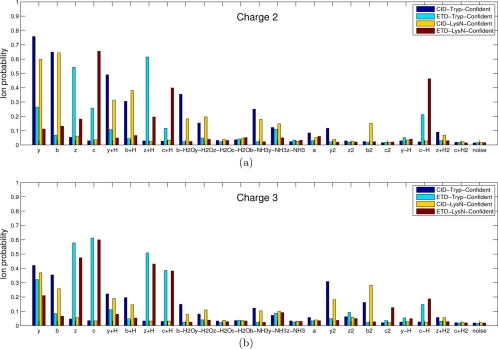
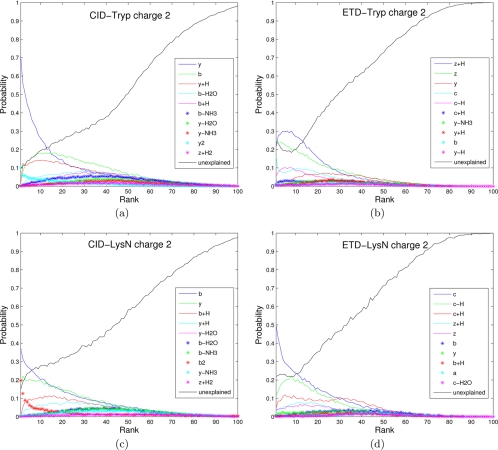
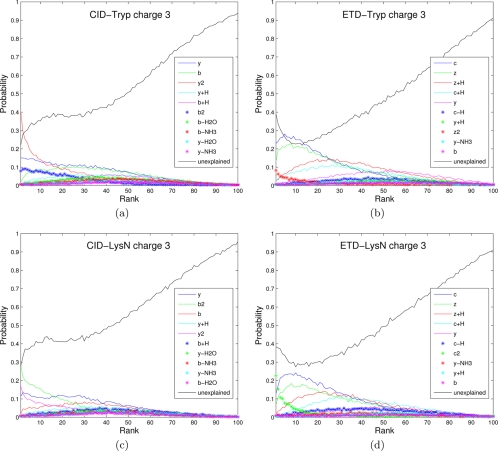
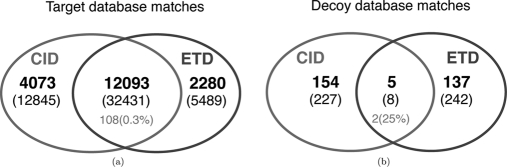
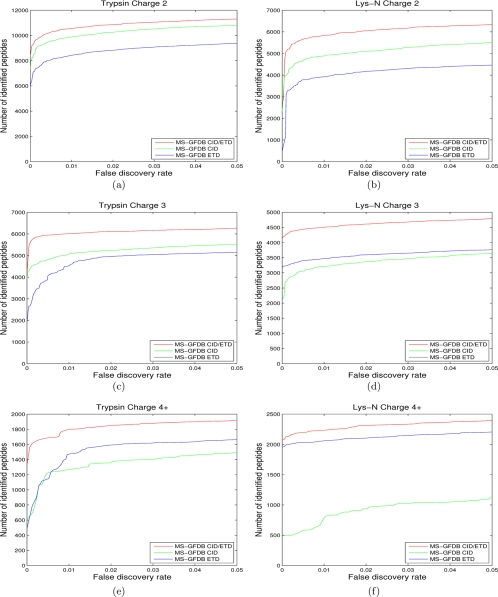
Recent emergence of new mass spectrometry techniques (e.g. electron transfer dissociation, ETD) and improved availability of additional proteases (e.g. Lys-N) for protein digestion in high-throughput experiments raised the challenge of designing new algorithms for interpreting the resulting new types of tandem mass (MS/MS) spectra. Traditional MS/MS database search algorithms such as SEQUEST and Mascot were originally designed for collision induced dissociation (CID) of tryptic peptides and are largely based on expert knowledge about fragmentation of tryptic peptides (rather than machine learning techniques) to design CID-specific scoring functions. As a result, the performance of these algorithms is suboptimal for new mass spectrometry technologies or nontryptic peptides. We recently proposed the generating function approach (MS-GF) for CID spectra of tryptic peptides. In this study, we extend MS-GF to automatically derive scoring parameters from a set of annotated MS/MS spectra of any type (e.g. CID, ETD, etc.), and present a new database search tool MS-GFDB based on MS-GF. We show that MS-GFDB outperforms Mascot for ETD spectra or peptides digested with Lys-N. For example, in the case of ETD spectra, the number of tryptic and Lys-N peptides identified by MS-GFDB increased by a factor of 2.7 and 2.6 as compared with Mascot. Moreover, even following a decade of Mascot developments for analyzing CID spectra of tryptic peptides, MS-GFDB (that is not particularly tailored for CID spectra or tryptic peptides) resulted in 28% increase over Mascot in the number of peptide identifications. Finally, we propose a statistical framework for analyzing multiple spectra from the same precursor (e.g. CID/ETD spectral pairs) and assigning p values to peptide-spectrum-spectrum matches.
Figures
References
-
- Zubarev R., Kelleher N., McLafferty F. (1998) Electron capture dissociation of multiply charged protein cations. a nonergodic process. J. Am. Chem. Soc. 120, 3265–3266
-
- Cooper H. J., Håkansson K., Marshall A. G. (2005) The role of electron capture dissociation in biomolecular analysis. Mass Spectrom. Rev. 24, 201–222 - PubMed
-
- Khidekel N., Ficarro S. B., Clark P. M., Bryan M. C., Swaney D. L., Rexach J. E., Sun Y. E., Coon J. J., Peters E. C., Hsieh-Wilson L. C. (2007) Probing the dynamics of o-glcnac glycosylation in the brain using quantitative proteomics. Nat. Chem. Biol. 3, 339–348 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
