

Targeting nonhomologous end-joining through epidermal growth factor receptor inhibition: rationale and strategies for radiosensitization
- PMID: 20832017
- PMCID: PMC2941735
- DOI: 10.1016/j.semradonc.2010.05.002
Targeting nonhomologous end-joining through epidermal growth factor receptor inhibition: rationale and strategies for radiosensitization
Abstract
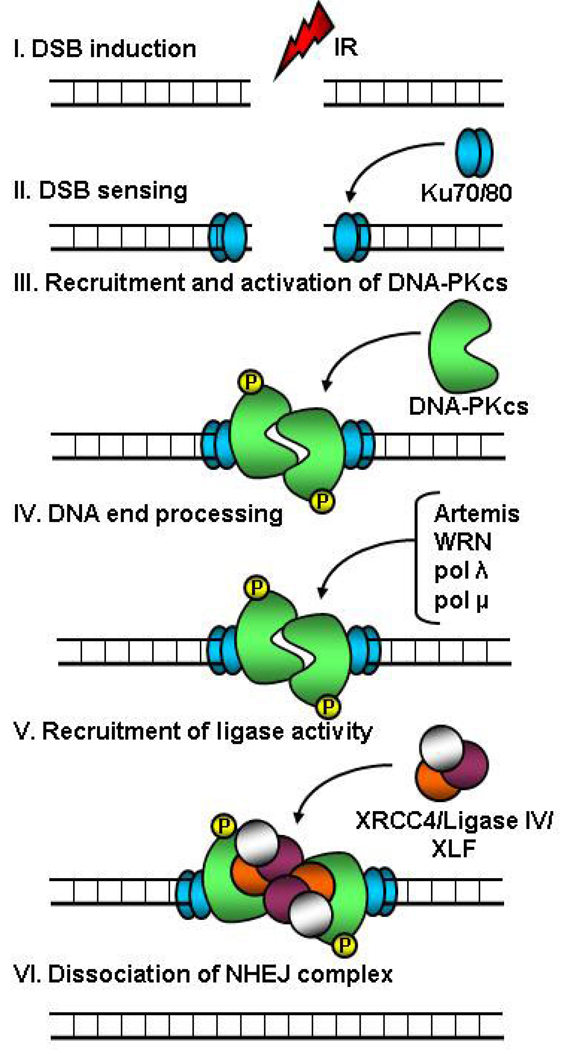
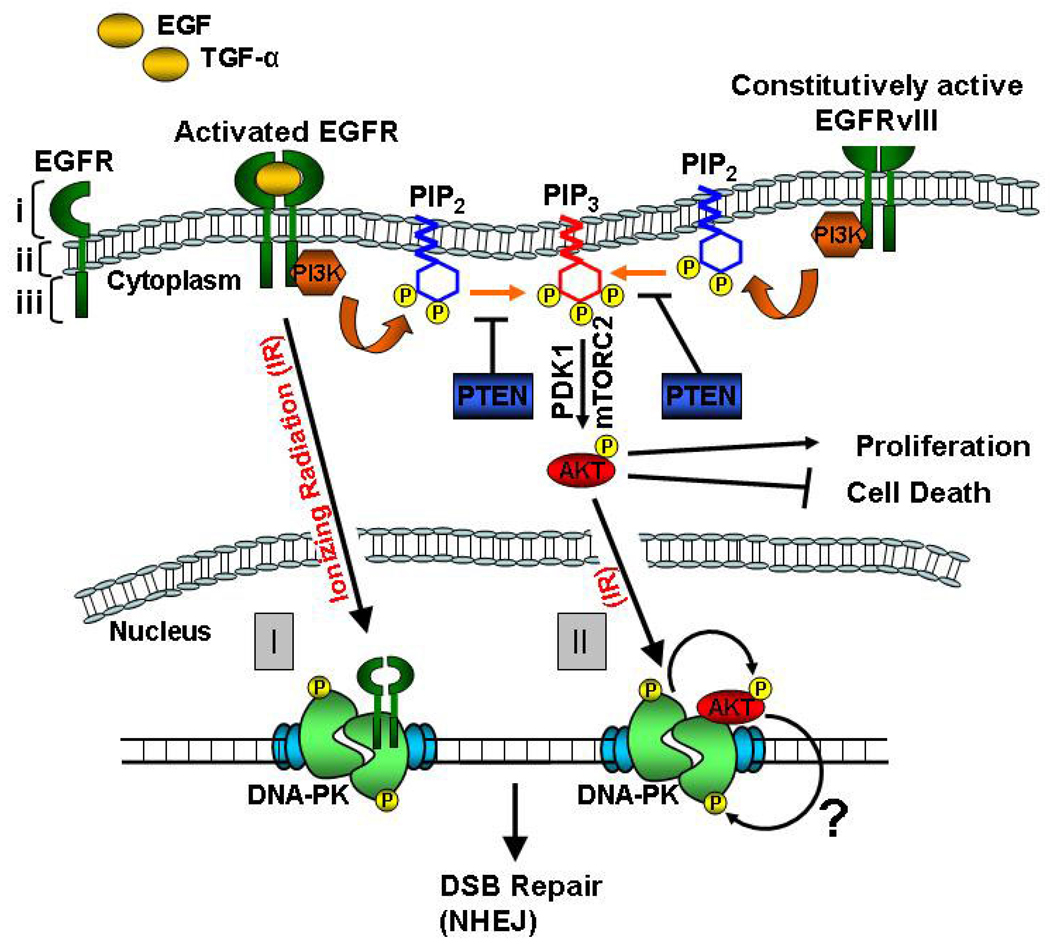
DNA double-strand breaks (DSBs) are the most lethal type of DNA damage induced by ionizing radiation or chemotherapeutic drugs used to eradicate cancer cells. The ability of cancer cells to effectively repair DSBs significantly influences the outcome of therapeutic regimens. Therefore, a new and important area of clinical cancer research is the development of DNA repair inhibitors that can be used as radio- or chemosensitizers. Nonhomologous end joining (NHEJ) is the predominant pathway for the repair of radiation-induced DSBs. A series of recent reports indicates that the epidermal growth factor receptor (EGFR) or its downstream components may modulate NHEJ through direct interaction with the DNA repair enzyme, DNA-dependent protein kinase. Because EGFR is overexpressed or activated in many cancers, these findings provide a compelling rationale for combining radiotherapy with therapies that block EGFR or its downstream signaling components. In this review, we delineate how these novel connections between a cell-surface receptor (EGFR) and a predominantly nuclear event (NHEJ) provide vulnerable nodes that can be selectively targeted to improve cancer therapy.
Copyright © 2010 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Enhancing radiotherapy through a greater understanding of homologous recombination.Semin Radiat Oncol. 2010 Oct;20(4):267-273.e3. doi: 10.1016/j.semradonc.2010.05.001. Semin Radiat Oncol. 2010. PMID: 20832019 Free PMC article. Review.
-
Poly(ADP-ribose) polymerase inhibition as a model for synthetic lethality in developing radiation oncology targets.Semin Radiat Oncol. 2010 Oct;20(4):274-81. doi: 10.1016/j.semradonc.2010.06.001. Semin Radiat Oncol. 2010. PMID: 20832020 Review.
-
Single-strand annealing, conservative homologous recombination, nonhomologous DNA end joining, and the cell cycle-dependent repair of DNA double-strand breaks induced by sparsely or densely ionizing radiation.Radiat Res. 2009 Mar;171(3):265-73. doi: 10.1667/RR0784.1. Radiat Res. 2009. PMID: 19267553
-
The role of nonhomologous DNA end joining, conservative homologous recombination, and single-strand annealing in the cell cycle-dependent repair of DNA double-strand breaks induced by H(2)O(2) in mammalian cells.Radiat Res. 2008 Dec;170(6):784-93. doi: 10.1667/RR1375.1. Radiat Res. 2008. PMID: 19138034
-
Targeting base excision repair as a sensitization strategy in radiotherapy.Semin Radiat Oncol. 2010 Oct;20(4):241-9. doi: 10.1016/j.semradonc.2010.05.005. Semin Radiat Oncol. 2010. PMID: 20832016 Review.
Cited by
-
Modulatory Role of microRNAs in Triple Negative Breast Cancer with Basal-Like Phenotype.Cancers (Basel). 2020 Nov 7;12(11):3298. doi: 10.3390/cancers12113298. Cancers (Basel). 2020. PMID: 33171872 Free PMC article. Review.
-
Influence of allelic Variations of hypoxia-related and DNA repair genes on patient outcome and toxicity in head and neck cancer treated with radiotherapy plus cetuximab.Eur Arch Otorhinolaryngol. 2016 Aug;273(8):2193-9. doi: 10.1007/s00405-015-3740-4. Epub 2015 Aug 6. Eur Arch Otorhinolaryngol. 2016. PMID: 26245169
-
Phosphorylation of TRIP13 at Y56 induces radiation resistance but sensitizes head and neck cancer to cetuximab.Mol Ther. 2022 Jan 5;30(1):468-484. doi: 10.1016/j.ymthe.2021.06.009. Epub 2021 Jun 8. Mol Ther. 2022. PMID: 34111559 Free PMC article.
-
Rationale-based therapeutic combinations with PI3K inhibitors in cancer treatment.Mol Cell Oncol. 2014 Oct 29;1(3):e963447. doi: 10.4161/23723548.2014.963447. eCollection 2014 Jul-Sep. Mol Cell Oncol. 2014. PMID: 27308344 Free PMC article. Review.
-
Regulatory mechanisms and clinical perspectives of miRNA in tumor radiosensitivity.Carcinogenesis. 2012 Nov;33(11):2220-7. doi: 10.1093/carcin/bgs235. Epub 2012 Jul 12. Carcinogenesis. 2012. PMID: 22798379 Free PMC article. Review.
References
-
- Martin SA, Lord CJ, Ashworth A. DNA repair deficiency as a therapeutic target in cancer. Curr Opin Genet Dev. 2008;18(1):80–86. - PubMed
-
- Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297(5578):63–64. - PubMed
-
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319(5868):1352–1355. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
