

Crystal structure of Lon protease: molecular architecture of gated entry to a sequestered degradation chamber
- PMID: 20834233
- PMCID: PMC2964173
- DOI: 10.1038/emboj.2010.226
Crystal structure of Lon protease: molecular architecture of gated entry to a sequestered degradation chamber
Abstract
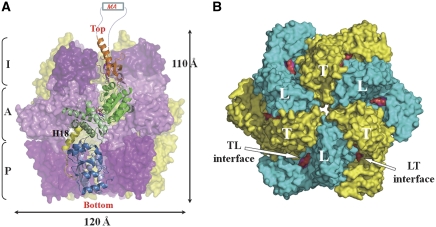
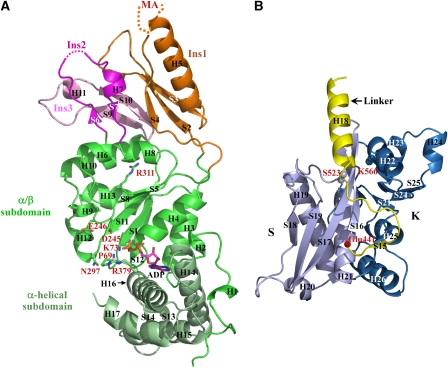
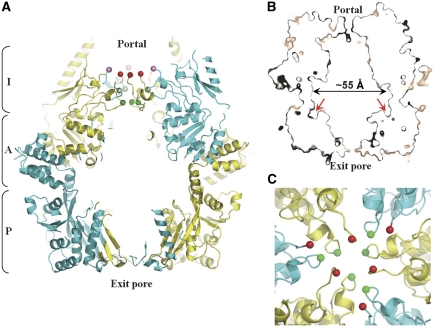
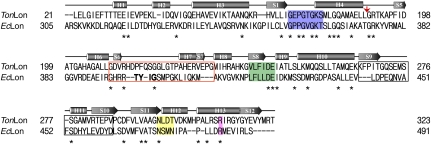
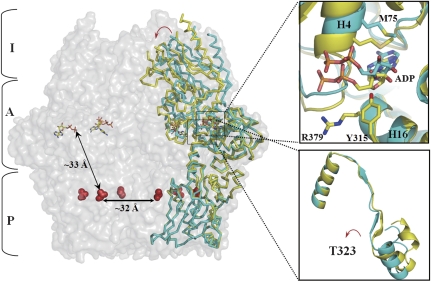
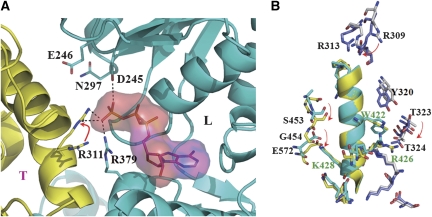
Lon proteases are distributed in all kingdoms of life and are required for survival of cells under stress. Lon is a tandem fusion of an AAA+ molecular chaperone and a protease with a serine-lysine catalytic dyad. We report the 2.0-Å resolution crystal structure of Thermococcus onnurineus NA1 Lon (TonLon). The structure is a three-tiered hexagonal cylinder with a large sequestered chamber accessible through an axial channel. Conserved loops extending from the AAA+ domain combine with an insertion domain containing the membrane anchor to form an apical domain that serves as a gate governing substrate access to an internal unfolding and degradation chamber. Alternating AAA+ domains are in tight- and weak-binding nucleotide states with different domain orientations and intersubunit contacts, reflecting intramolecular dynamics during ATP-driven protein unfolding and translocation. The bowl-shaped proteolytic chamber is contiguous with the chaperone chamber allowing internalized proteins direct access to the proteolytic sites without further gating restrictions.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Baumeister W, Walz J, Zuhl F, Seemuller E (1998) The proteasome: paradigm of a self-compartmentalizing protease. Cell 92: 367–380 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
