

Algorithmic and analytical methods in network biology
- PMID: 20836029
- PMCID: PMC3087298
- DOI: 10.1002/wsbm.61
Algorithmic and analytical methods in network biology
Abstract
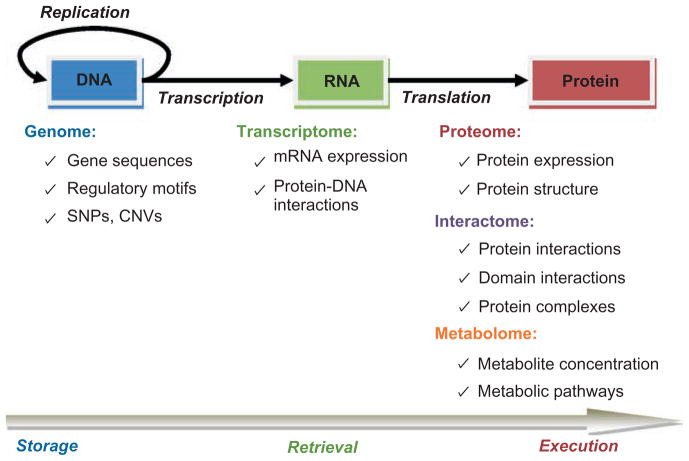
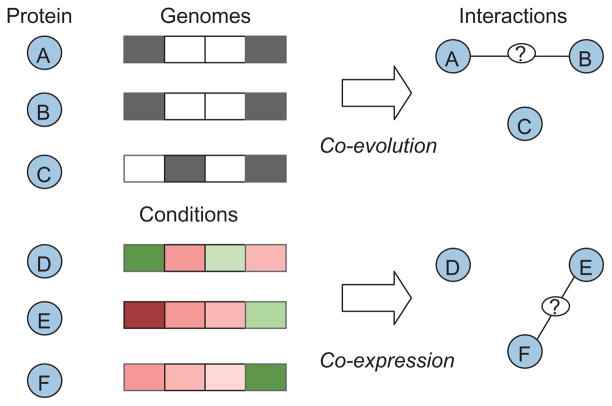
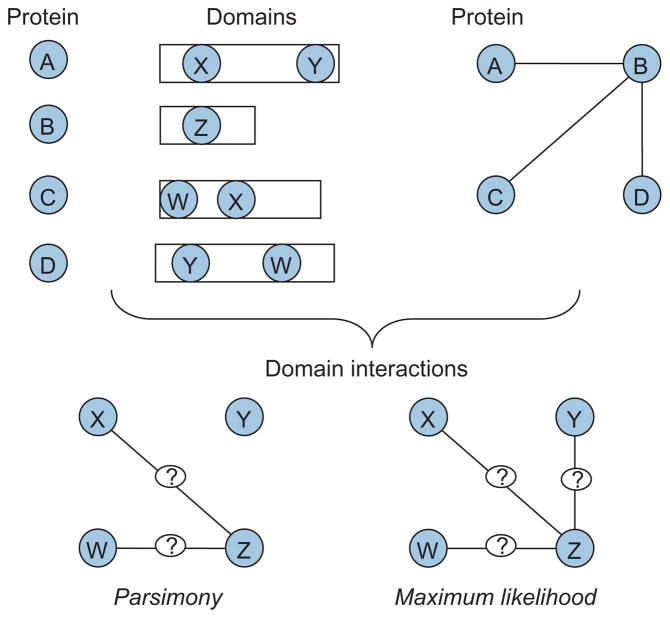
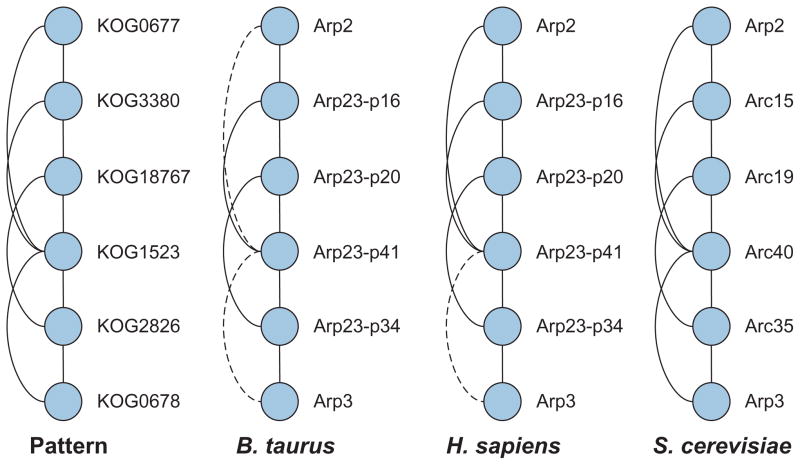
During the genomic revolution, algorithmic and analytical methods for organizing, integrating, analyzing, and querying biological sequence data proved invaluable. Today, increasing availability of high-throughput data pertaining to functional states of biomolecules, as well as their interactions, enables genome-scale studies of the cell from a systems perspective. The past decade witnessed significant efforts on the development of computational infrastructure for large-scale modeling and analysis of biological systems, commonly using network models. Such efforts lead to novel insights into the complexity of living systems, through development of sophisticated abstractions, algorithms, and analytical techniques that address a broad range of problems, including the following: (1) inference and reconstruction of complex cellular networks; (2) identification of common and coherent patterns in cellular networks, with a view to understanding the organizing principles and building blocks of cellular signaling, regulation, and metabolism; and (3) characterization of cellular mechanisms that underlie the differences between living systems, in terms of evolutionary diversity, development and differentiation, and complex phenotypes, including human disease. These problems pose significant algorithmic and analytical challenges because of the inherent complexity of the systems being studied; limitations of data in terms of availability, scope, and scale; intractability of resulting computational problems; and limitations of reference models for reliable statistical inference. This article provides a broad overview of existing algorithmic and analytical approaches to these problems, highlights key biological insights provided by these approaches, and outlines emerging opportunities and challenges in computational systems biology.
Figures
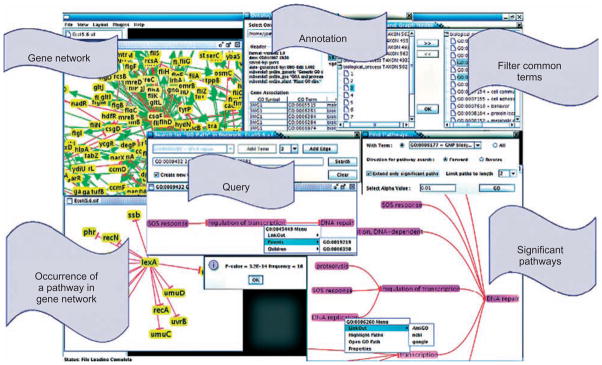
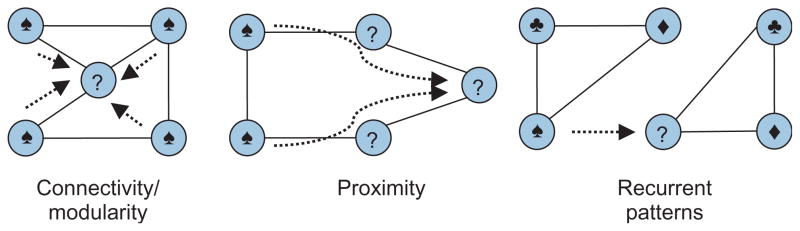
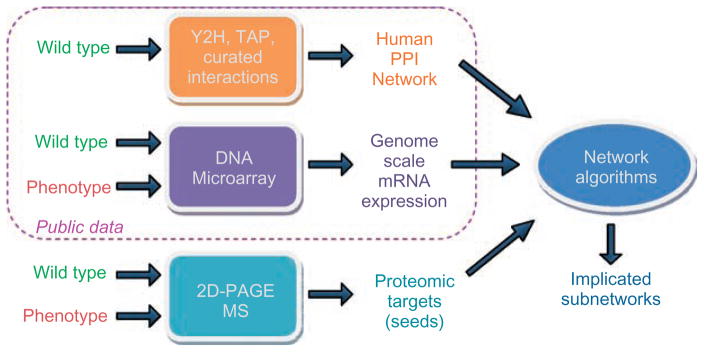
Similar articles
-
Network integration and graph analysis in mammalian molecular systems biology.IET Syst Biol. 2008 Sep;2(5):206-21. doi: 10.1049/iet-syb:20070075. IET Syst Biol. 2008. PMID: 19045817 Free PMC article. Review.
-
Gene regulatory network inference: data integration in dynamic models-a review.Biosystems. 2009 Apr;96(1):86-103. doi: 10.1016/j.biosystems.2008.12.004. Epub 2008 Dec 27. Biosystems. 2009. PMID: 19150482 Review.
-
The application of flux balance analysis in systems biology.Wiley Interdiscip Rev Syst Biol Med. 2010 May-Jun;2(3):372-382. doi: 10.1002/wsbm.60. Wiley Interdiscip Rev Syst Biol Med. 2010. PMID: 20836035 Review.
-
Building the developmental oculome: systems biology in vertebrate eye development and disease.Wiley Interdiscip Rev Syst Biol Med. 2010 May-Jun;2(3):305-323. doi: 10.1002/wsbm.59. Wiley Interdiscip Rev Syst Biol Med. 2010. PMID: 20836031 Free PMC article. Review.
-
Network legos: building blocks of cellular wiring diagrams.J Comput Biol. 2008 Sep;15(7):829-44. doi: 10.1089/cmb.2007.0139. J Comput Biol. 2008. PMID: 18707557
Cited by
-
Structure-based systems biology for analyzing off-target binding.Curr Opin Struct Biol. 2011 Apr;21(2):189-99. doi: 10.1016/j.sbi.2011.01.004. Epub 2011 Feb 1. Curr Opin Struct Biol. 2011. PMID: 21292475 Free PMC article. Review.
-
Augmentation of crop productivity through interventions of omics technologies in India: challenges and opportunities.3 Biotech. 2018 Nov;8(11):454. doi: 10.1007/s13205-018-1473-y. Epub 2018 Oct 19. 3 Biotech. 2018. PMID: 30370195 Free PMC article. Review.
-
A network-based approach to classify the three domains of life.Biol Direct. 2011 Oct 13;6:53. doi: 10.1186/1745-6150-6-53. Biol Direct. 2011. PMID: 21995640 Free PMC article.
-
Identification of rifampin-regulated functional modules and related microRNAs in human hepatocytes based on the protein interaction network.BMC Genomics. 2016 Aug 22;17 Suppl 7(Suppl 7):517. doi: 10.1186/s12864-016-2909-6. BMC Genomics. 2016. PMID: 27557147 Free PMC article.
-
Evaluating the predictive accuracy of curated biological pathways in a public knowledgebase.Database (Oxford). 2022 Mar 28;2022:baac009. doi: 10.1093/database/baac009. Database (Oxford). 2022. PMID: 35348650 Free PMC article.
References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
-
- Larkin MA, Blackshields G, Brown NP, Chenna R, Mcgettigan PA, Mcwilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. - PubMed
-
- Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–1664. - PubMed
-
- Wang DG, Fan JB, Siao CJ, Berno A, Young P, et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science. 1998;280:1077–1082. - PubMed
-
- Pollack JR, Perou CM, Alizadeh AA, Eisen MB, Pergamenschikov A, et al. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet. 1999;23:41–46. - PubMed
FURTHER READING
-
- Barabasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5:101–113. - PubMed
-
- Joyce AR, Palsson BØ. The model organism as a system: integrating ‘omics’ data sets. Nat Rev Mol Cell Biol. 2006;7:198–210. - PubMed
-
- Bornholdt S. Less is more in modelling large genetic networks. Science. 2005;310:449–451. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
