

Genes that determine immunology and inflammation modify the basic defect of impaired ion conductance in cystic fibrosis epithelia
- PMID: 20837493
- PMCID: PMC3003880
- DOI: 10.1136/jmg.2010.080937
Genes that determine immunology and inflammation modify the basic defect of impaired ion conductance in cystic fibrosis epithelia
Abstract
Background: The cystic fibrosis (CF) basic defect, caused by dysfunction of the apical chloride channel CFTR in the gastrointestinal and respiratory tract epithelia, has not been employed so far to support the role of CF modifier genes.
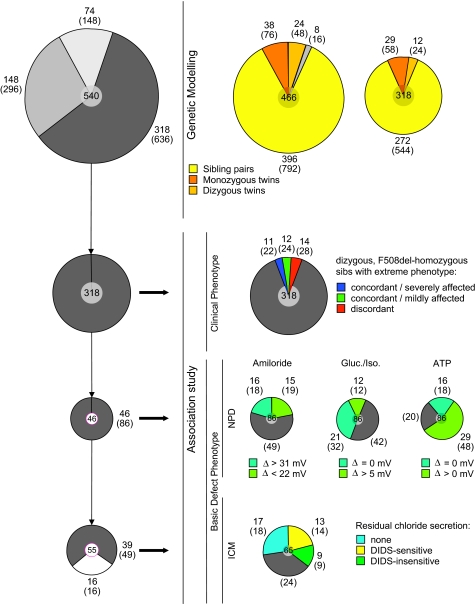
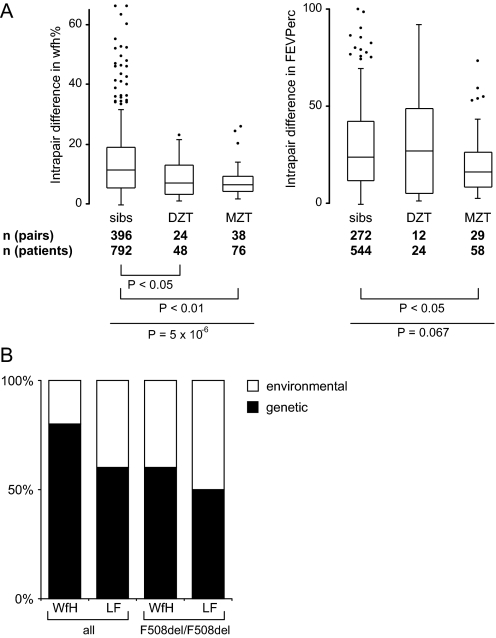
Methods: Patients were selected from 101 families with a total of 171 F508del-CFTR homozygous CF patients to identify CF modifying genes. A candidate gene based association study of 52 genes on 16 different chromosomes with a total of 182 genetic markers was performed. Differences in haplotype and/or diplotype distribution between case and reference CF subpopulations were analysed.
Results: Variants at immunologically relevant genes were associated with the manifestation of the CF basic defect (0.01<Praw<0.0001 at IL1B, TLR9, TNFα, CD95, STAT3 and TNFR). The intragenic background of F508del-CFTR chromosomes determined disease severity and manifestation of the basic defect (Praw=0.0009). Allele distributions comparing transmitted and non-transmitted alleles were distorted at several loci unlinked to CFTR.
Conclusions: The inherited capabilities of the innate and adaptive immune system determine the manifestation of the CF basic defect. Variants on F508del-CFTR chromosomes contribute to the observed patient-to-patient variability among F508del-CFTR homozygotes. A survivor effect, manifesting as a transmission disequilibrium at many loci, is consistent with the improvement of clinical care over the last decades, resulting in a depletion of risk alleles at modifier genes. Awareness of non-genetic factors such as improvement of patient care over time is crucial for the interpretation of CF modifier studies.
Conflict of interest statement
Figures


References
-
- Strausbaugh SD, Davis PB. Cystic fibrosis: a review of epidemiology and pathobiology. Clin Chest Med 2007;28:279–88 - PubMed
-
- Koch C, Cuppens H, Rainisio M, Madessani U, Harms H, Hodson M, Mastella G, Navarro J, Strandvik B, McKenzie S. Investigators of the ERCF. European Epidemiologic Registry of Cystic Fibrosis (ERCF): comparison of major disease manifestations between patients with different classes of mutations. Pediatr Pulmonol 2001;31:1–12 - PubMed
-
- Mekus F, Ballmann M, Bronsveld I, Bijman J, Veeze H, Tümmler B. Categories of deltaF508 homozygous cystic fibrosis twin and sibling pairs with distinct phenotypic characteristics. Twin Res 2000;3:277–93 - PubMed
-
- Davis PB. Cystic fibrosis since 1938. Am J Respir Crit Care Med 2006;173:475–82 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous