

Tackling the widespread and critical impact of batch effects in high-throughput data
- PMID: 20838408
- PMCID: PMC3880143
- DOI: 10.1038/nrg2825
Tackling the widespread and critical impact of batch effects in high-throughput data
Abstract
High-throughput technologies are widely used, for example to assay genetic variants, gene and protein expression, and epigenetic modifications. One often overlooked complication with such studies is batch effects, which occur because measurements are affected by laboratory conditions, reagent lots and personnel differences. This becomes a major problem when batch effects are correlated with an outcome of interest and lead to incorrect conclusions. Using both published studies and our own analyses, we argue that batch effects (as well as other technical and biological artefacts) are widespread and critical to address. We review experimental and computational approaches for doing so.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
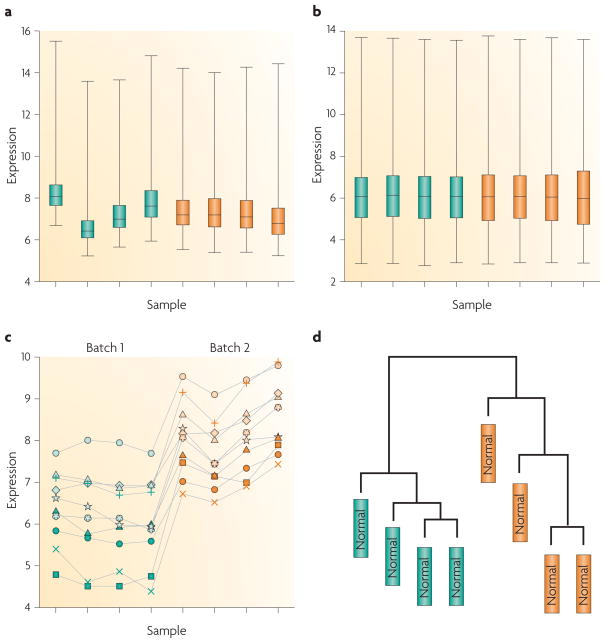
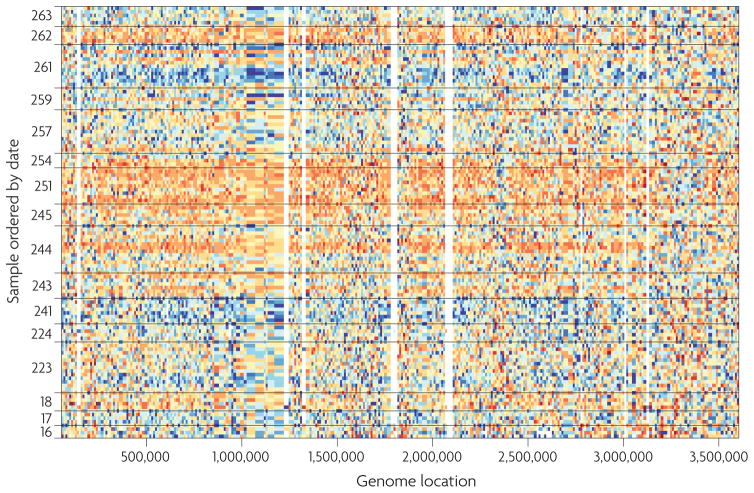
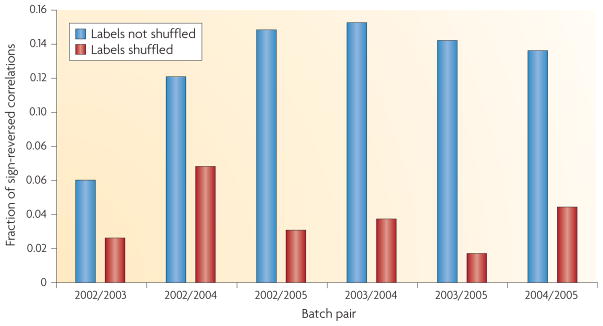
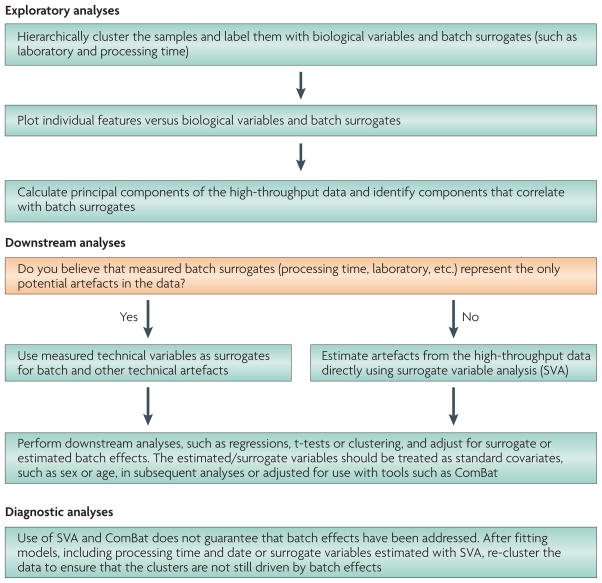
References
-
- Youden WJ. Enduring values. Technometrics. 1972;14:1–11.
-
- Petricoin EF, et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 2002;359:572–577. - PubMed
-
- Akey JM, Biswas S, Leek JT, Storey JD. On the design and analysis of gene expression studies in human populations. Nature Genet. 2007;39:807–808. author reply 808–809. - PubMed
-
- Baggerly KA, Edmonson SR, Morris JS, Coombes KR. High-resolution serum proteomic patterns for ovarian cancer detection. Endocr Relat Cancer. 2004;11:583–584. author reply 585–587. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
