

Skeletal muscle in motor neuron diseases: therapeutic target and delivery route for potential treatments
- PMID: 20840067
- PMCID: PMC4834127
- DOI: 10.2174/1389450111007011250
Skeletal muscle in motor neuron diseases: therapeutic target and delivery route for potential treatments
Abstract
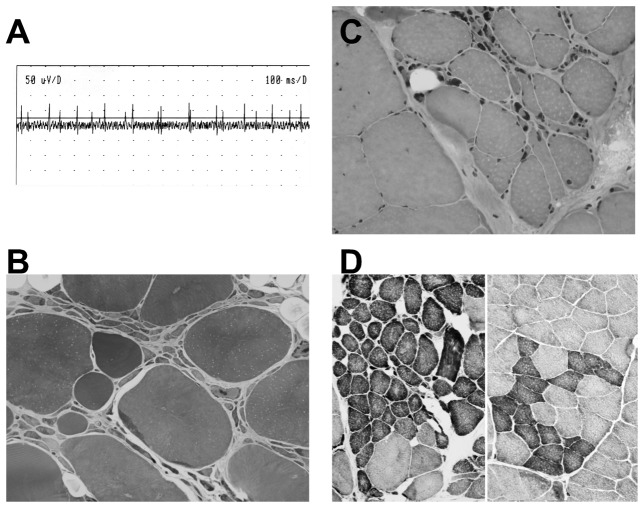
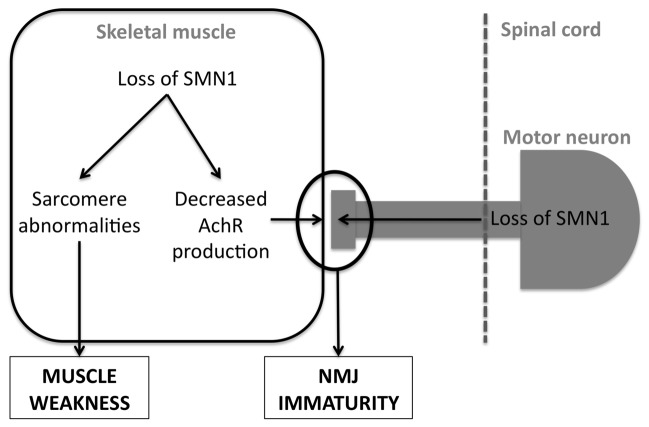
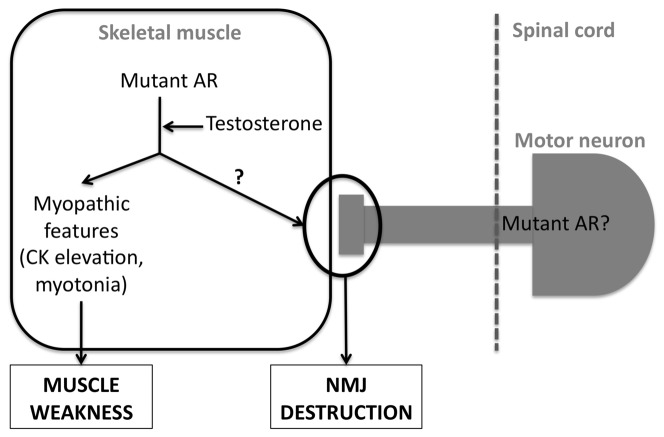
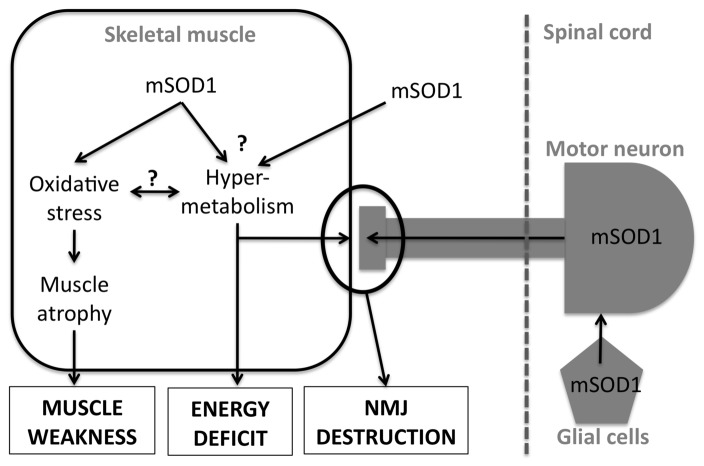
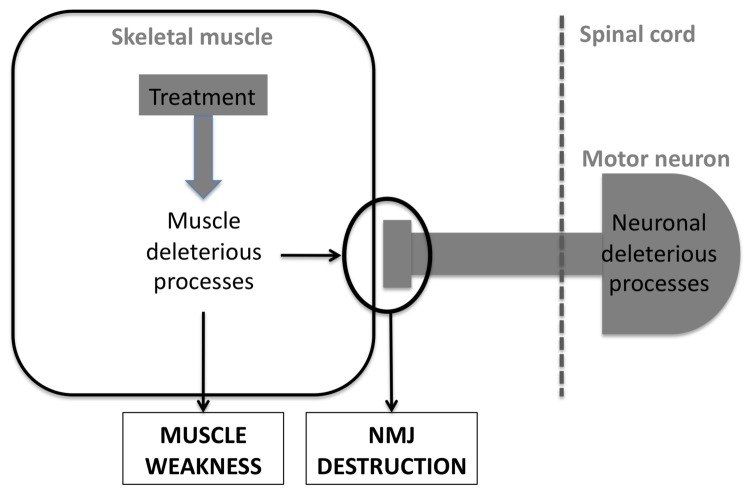
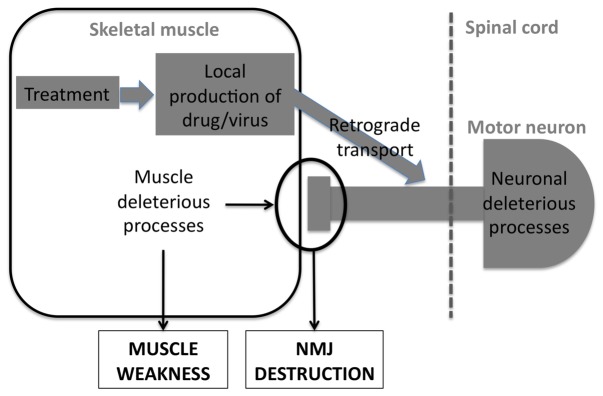
Lower motor neuron (LMN) degeneration occurs in several diseases that affect patients from neonates to elderly and can either be genetically transmitted or occur sporadically. Among diseases involving LMN degeneration, spinal muscular atrophy (SMA) and spinal bulbar muscular atrophy (Kennedy's disease, SBMA) are pure genetic diseases linked to loss of the SMN gene (SMA) or expansion of a polyglutamine tract in the androgen receptor gene (SBMA) while amyotrophic lateral sclerosis (ALS) can either be of genetic origin or occur sporadically. In this review, our aim is to put forward the hypothesis that muscle fiber atrophy and weakness might not be a simple collateral damage of LMN degeneration, but instead that muscle fibers may be the site of crucial pathogenic events in these diseases. In SMA, the SMN gene was shown to be required for muscle structure and strength as well as for neuromuscular junction formation, and a subset of SMA patients develop myopathic pathology. In SBMA, the occurrence of myopathic histopathology in patients and animal models, along with neuromuscular phenotype of animal models expressing the androgen receptor in muscle only has lead to the proposal that SBMA may indeed be a muscle disease. Lastly, in ALS, at least part of the phenotype might be explained by pathogenic events occuring in skeletal muscle. Apart from its potential pathogenic role, skeletal muscle pathophysiological events might be a target for treatments and/or be a preferential route for targeting motor neurons.
Figures
References
-
- Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120–2133. - PubMed
-
- Katsuno M, Adachi H, Waza M, Banno H, Suzuki K, Tanaka F, Doyu M, Sobue G. Pathogenesis, animal models and therapeutics in spinal and bulbar muscular atrophy (sbma) Exp Neurol. 2006;200:8–18. - PubMed
-
- Brooks BR, Sanjak M, Belden D, Juhasz-Poscine K, Waclawik A. Natural history of amyotrophic lateral sclerosis. In: Brown RHJ, Meininger V, Swash M, editors. Amyotrophic lateral sclerosis. London: Dunitz; 2000. pp. 31–58. - PubMed
-
- Baloh RH, Rakowicz W, Gardner R, Pestronk A. Frequent atrophic groups with mixed-type myofibers is distinctive to motor neuron syndromes. Muscle Nerve. 2007;36:107–110. - PubMed
-
- Henderson CE, Bloch-Gallego E, Camu W, Gouin A, Lemeulle C, Mettling C. Motoneuron survival factors: Biological roles and therapeutic potential. Neuromuscul Disord. 1993;3:455–458. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous