

Overlapping codes within protein-coding sequences
- PMID: 20841429
- PMCID: PMC2963821
- DOI: 10.1101/gr.105072.110
Overlapping codes within protein-coding sequences
Abstract
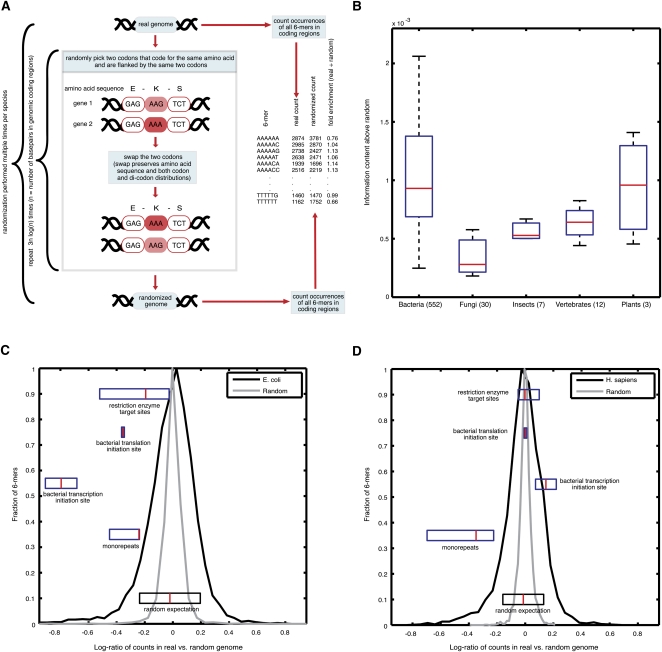
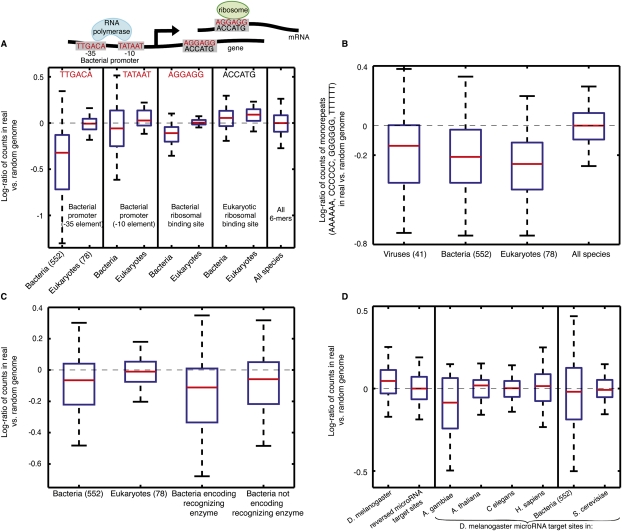
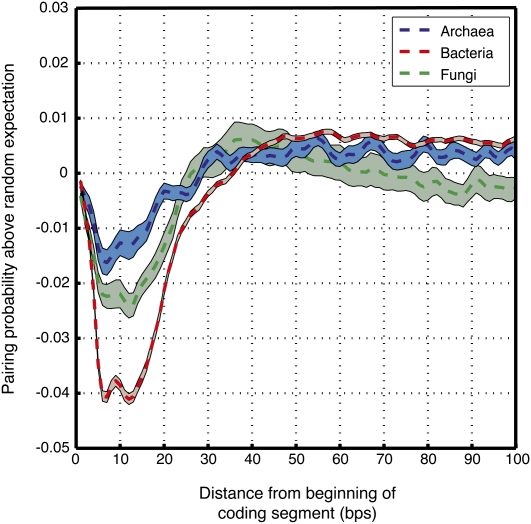
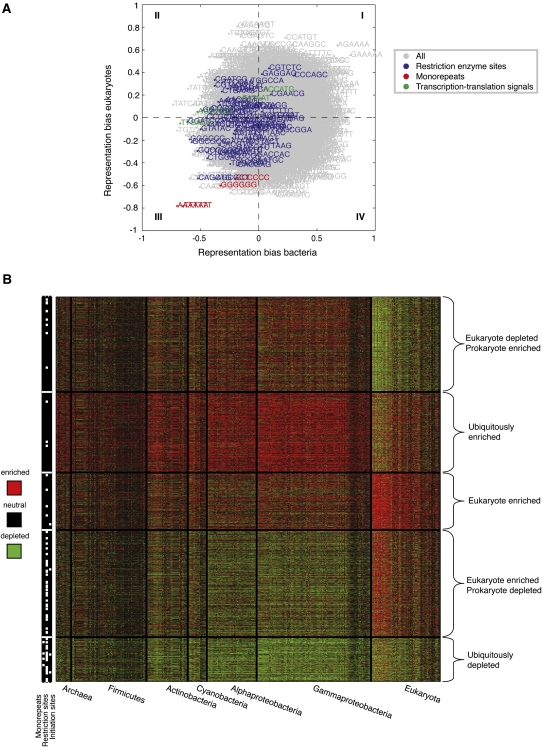
Genomes encode multiple signals, raising the question of how these different codes are organized along the linear genome sequence. Within protein-coding regions, the redundancy of the genetic code can, in principle, allow for the overlapping encoding of signals in addition to the amino acid sequence, but it is not known to what extent genomes exploit this potential and, if so, for what purpose. Here, we systematically explore whether protein-coding regions accommodate overlapping codes, by comparing the number of occurrences of each possible short sequence within the protein-coding regions of over 700 species from viruses to plants, to the same number in randomizations that preserve amino acid sequence and codon bias. We find that coding regions across all phyla encode additional information, with bacteria carrying more information than eukaryotes. The detailed signals consist of both known and potentially novel codes, including position-dependent secondary RNA structure, bacteria-specific depletion of transcription and translation initiation signals, and eukaryote-specific enrichment of microRNA target sites. Our results suggest that genomes may have evolved to encode extensive overlapping information within protein-coding regions.
Figures
Comment in
-
Biochemistry. Hidden code in the protein code.Nat Methods. 2010 Nov;7(11):874. doi: 10.1038/nmeth1110-874. Nat Methods. 2010. PMID: 21049579
References
-
- Bartel DP, Chen CZ 2004. Micromanagers of gene expression: The potentially widespread influence of metazoan microRNAs. Nat Rev Genet 5: 396–400 - PubMed
-
- Boycheva S, Chkodrov G, Ivanov I 2003. Codon pairs in the genome of Escherichia coli. Bioinformatics 19: 987–998 - PubMed
-
- Burge CB, Karlin S 1998. Finding the genes in genomic DNA. Curr Opin Struct Biol 8: 346–354 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources