

Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders
- PMID: 20842175
- PMCID: PMC2948541
- DOI: 10.1038/nrn2911
Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders
Abstract
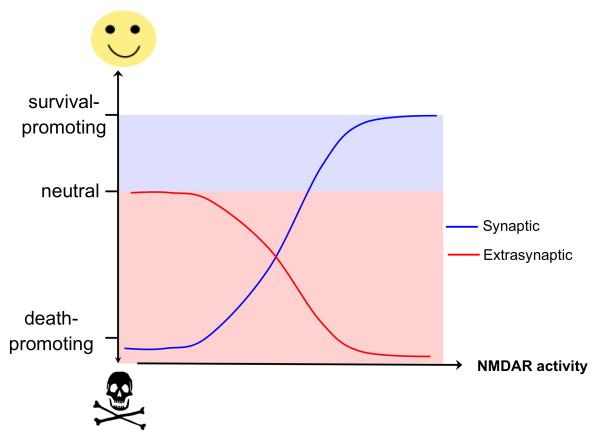
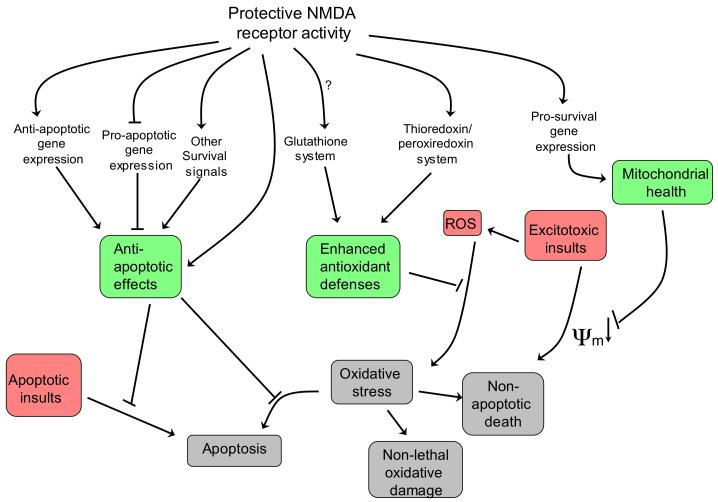
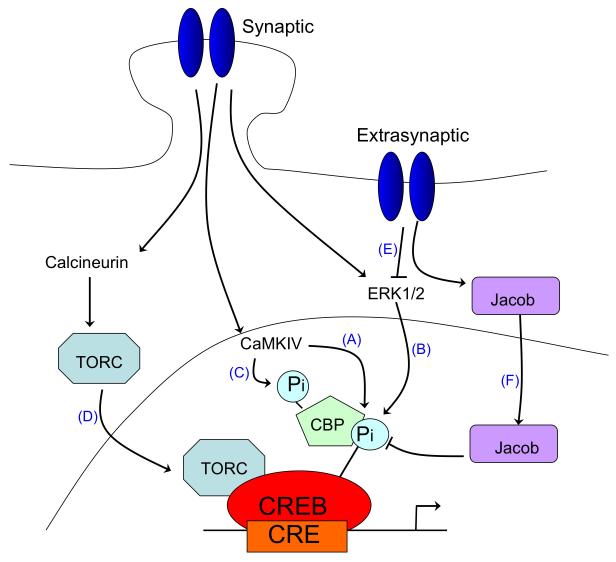
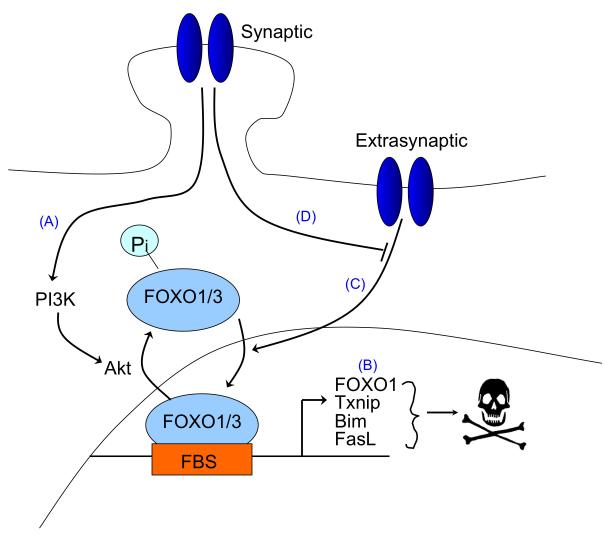
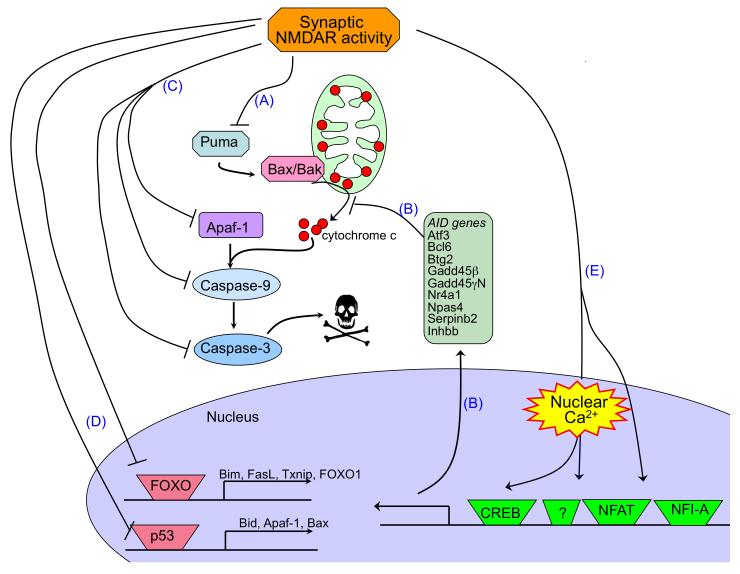
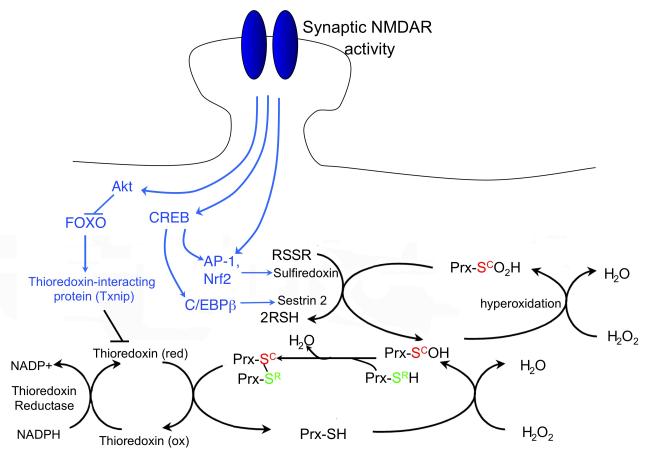
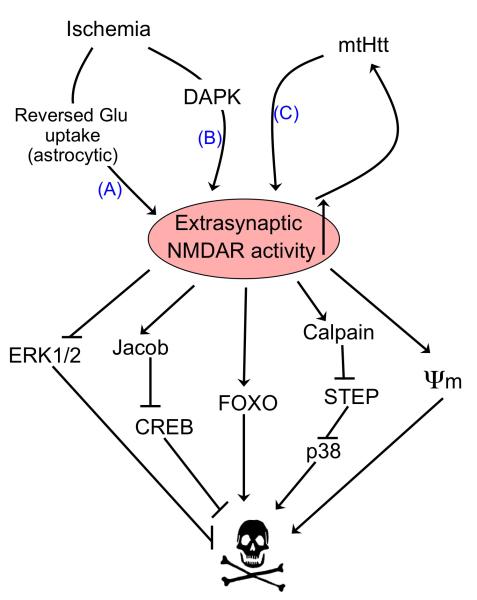
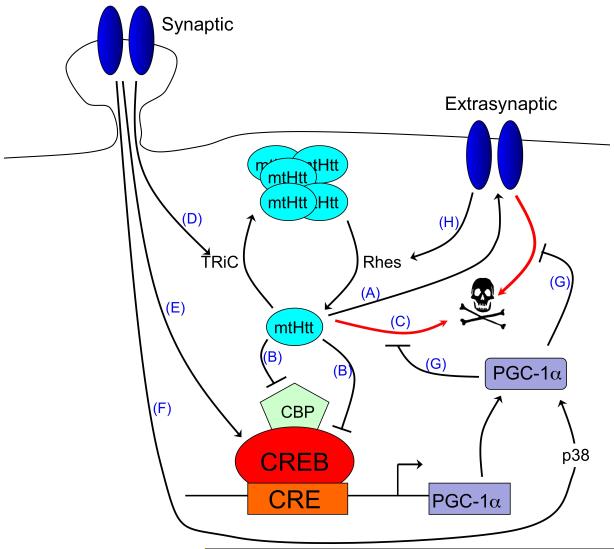
There is a long-standing paradox that NMDA (N-methyl-D-aspartate) receptors (NMDARs) can both promote neuronal health and kill neurons. Recent studies show that NMDAR-induced responses depend on the receptor location: stimulation of synaptic NMDARs, acting primarily through nuclear Ca(2+) signalling, leads to the build-up of a neuroprotective 'shield', whereas stimulation of extrasynaptic NMDARs promotes cell death. These differences result from the activation of distinct genomic programmes and from opposing actions on intracellular signalling pathways. Perturbations in the balance between synaptic and extrasynaptic NMDAR activity contribute to neuronal dysfunction in acute ischaemia and Huntington's disease, and could be a common theme in the aetiology of neurodegenerative diseases. Neuroprotective therapies should aim to both enhance the effect of synaptic activity and disrupt extrasynaptic NMDAR-dependent death signalling.
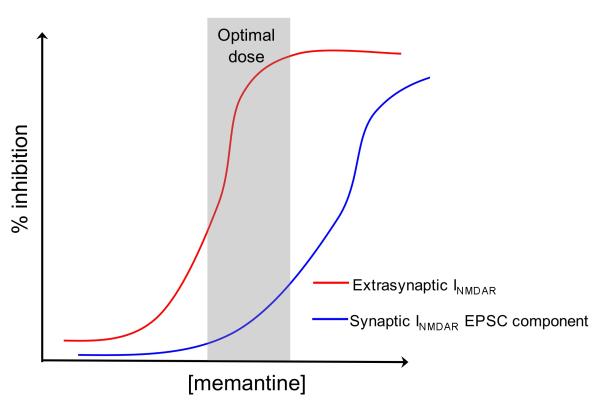
Figures
References
-
- Aamodt SM, Constantine-Paton M. The role of neural activity in synaptic development and its implications for adult brain function. Adv Neurol. 1999;79:133–44. - PubMed
-
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–9. - PubMed
-
- Cottrell JR, Dube GR, Egles C, Liu G. Distribution, density, and clustering of functional glutamate receptors before and after synaptogenesis in hippocampal neurons. J Neurophysiol. 2000;84:1573–87. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
