

Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia
- PMID: 20844535
- PMCID: PMC3355472
- DOI: 10.1038/nature09328
Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia
Abstract
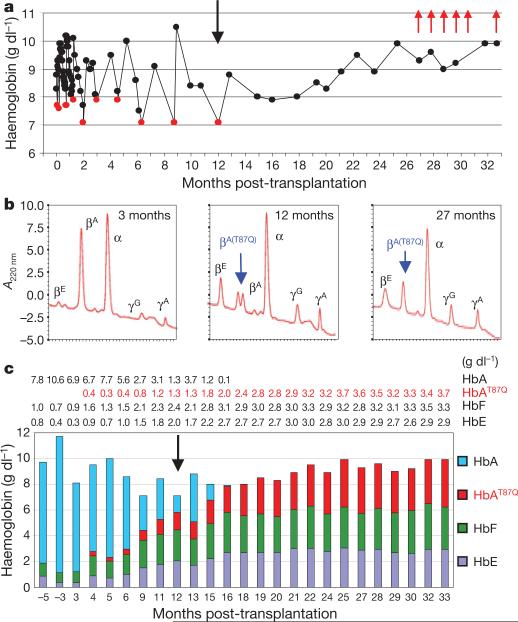
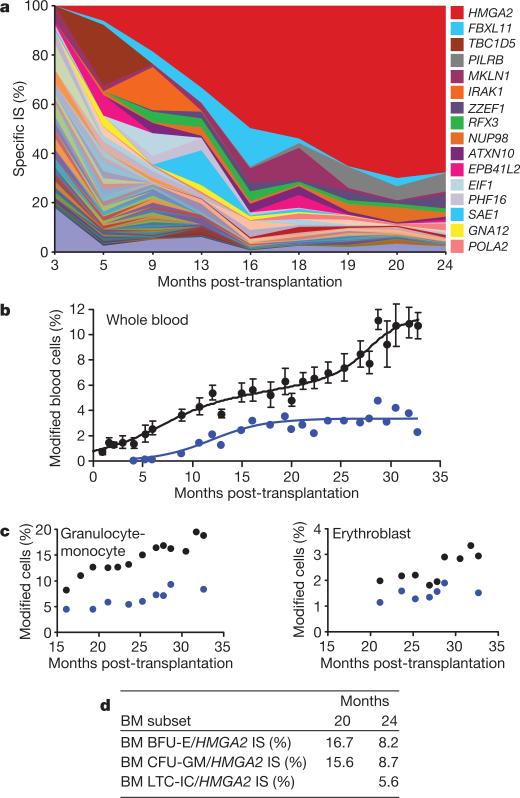
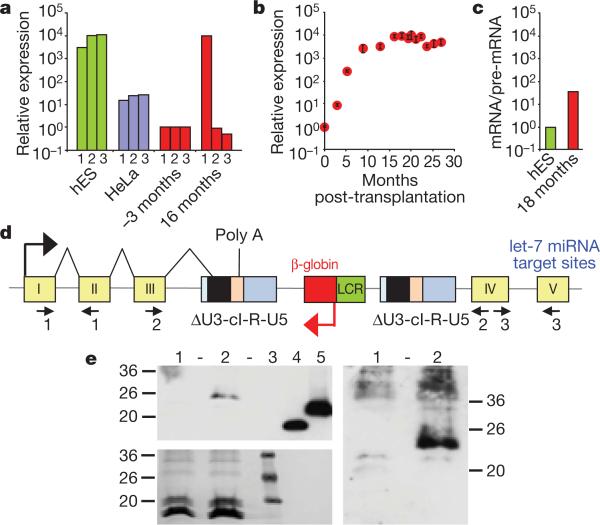
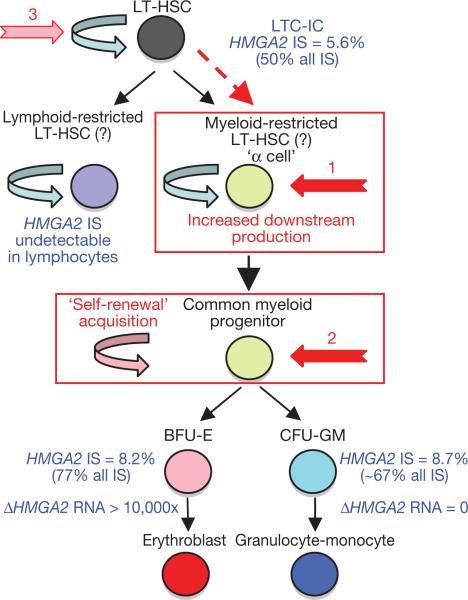
The β-haemoglobinopathies are the most prevalent inherited disorders worldwide. Gene therapy of β-thalassaemia is particularly challenging given the requirement for massive haemoglobin production in a lineage-specific manner and the lack of selective advantage for corrected haematopoietic stem cells. Compound β(E)/β(0)-thalassaemia is the most common form of severe thalassaemia in southeast Asian countries and their diasporas. The β(E)-globin allele bears a point mutation that causes alternative splicing. The abnormally spliced form is non-coding, whereas the correctly spliced messenger RNA expresses a mutated β(E)-globin with partial instability. When this is compounded with a non-functional β(0) allele, a profound decrease in β-globin synthesis results, and approximately half of β(E)/β(0)-thalassaemia patients are transfusion-dependent. The only available curative therapy is allogeneic haematopoietic stem cell transplantation, although most patients do not have a human-leukocyte-antigen-matched, geno-identical donor, and those who do still risk rejection or graft-versus-host disease. Here we show that, 33 months after lentiviral β-globin gene transfer, an adult patient with severe β(E)/β(0)-thalassaemia dependent on monthly transfusions since early childhood has become transfusion independent for the past 21 months. Blood haemoglobin is maintained between 9 and 10 g dl(-1), of which one-third contains vector-encoded β-globin. Most of the therapeutic benefit results from a dominant, myeloid-biased cell clone, in which the integrated vector causes transcriptional activation of HMGA2 in erythroid cells with further increased expression of a truncated HMGA2 mRNA insensitive to degradation by let-7 microRNAs. The clonal dominance that accompanies therapeutic efficacy may be coincidental and stochastic or result from a hitherto benign cell expansion caused by dysregulation of the HMGA2 gene in stem/progenitor cells.
Figures
Comment in
-
Gene therapy: Targeting β-thalassaemia.Nature. 2010 Sep 16;467(7313):277-8. doi: 10.1038/467277a. Nature. 2010. PMID: 20844523 No abstract available.
References
-
- Fucharoen S, Winichagoon P. Clinical and hematologic aspects of hemoglobin E β-thalassemia. Curr. Opin. Hematol. 2000;7:106–112. - PubMed
-
- Olivieri NF, et al. Studies in haemoglobin E β-thalassaemia. Br. J. Haematol. 2008;141:388–397. - PubMed
-
- Grosveld F, van Assendelft GB, Greaves DR, Kollias G. Position-independent, high-level expression of the human β-globin gene in transgenic mice. Cell. 1987;51:975–985. - PubMed
-
- May C, et al. Therapeutic haemoglobin synthesis in β-thalassaemic mice expressing lentivirus-encoded human β-globin. Nature. 2000;406:82–86. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
