

Quantitative analysis of a deeply sequenced marine microbial metatranscriptome
- PMID: 20844569
- PMCID: PMC3105723
- DOI: 10.1038/ismej.2010.141
Quantitative analysis of a deeply sequenced marine microbial metatranscriptome
Abstract
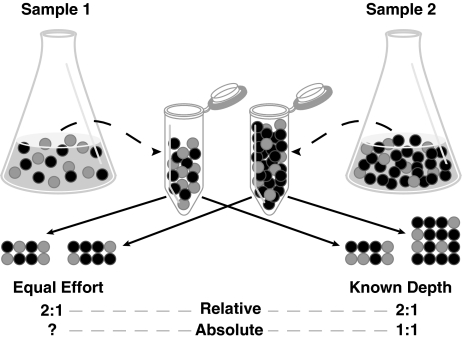
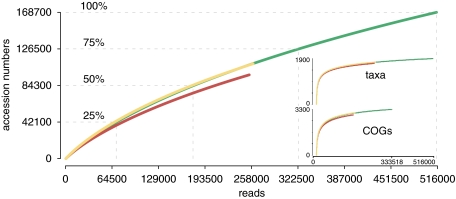
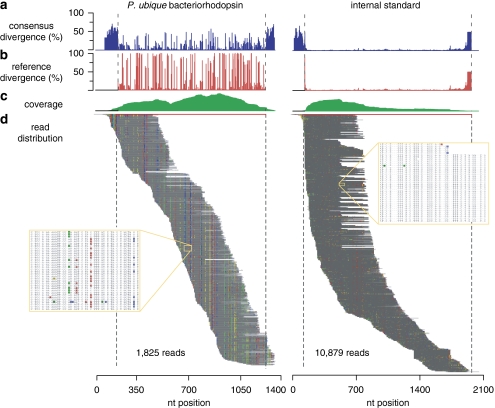
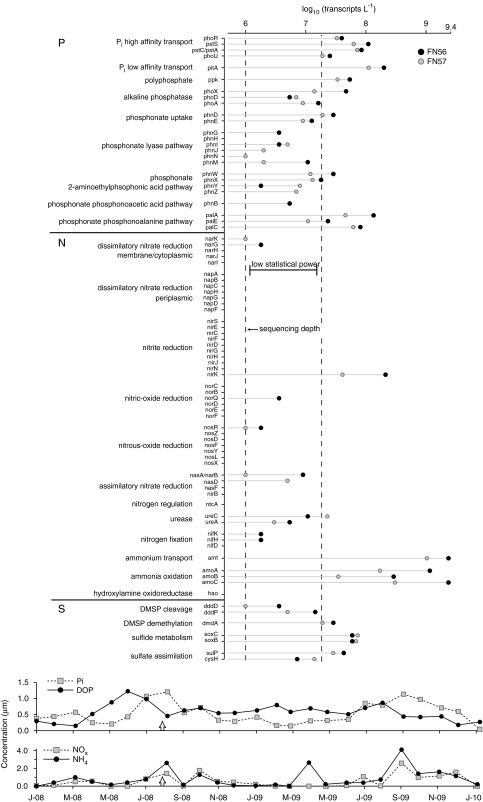
The potential of metatranscriptomic sequencing to provide insights into the environmental factors that regulate microbial activities depends on how fully the sequence libraries capture community expression (that is, sample-sequencing depth and coverage depth), and the sensitivity with which expression differences between communities can be detected (that is, statistical power for hypothesis testing). In this study, we use an internal standard approach to make absolute (per liter) estimates of transcript numbers, a significant advantage over proportional estimates that can be biased by expression changes in unrelated genes. Coastal waters of the southeastern United States contain 1 × 10(12) bacterioplankton mRNA molecules per liter of seawater (~200 mRNA molecules per bacterial cell). Even for the large bacterioplankton libraries obtained in this study (~500,000 possible protein-encoding sequences in each of two libraries after discarding rRNAs and small RNAs from >1 million 454 FLX pyrosequencing reads), sample-sequencing depth was only 0.00001%. Expression levels of 82 genes diagnostic for transformations in the marine nitrogen, phosphorus and sulfur cycles ranged from below detection (<1 × 10(6) transcripts per liter) for 36 genes (for example, phosphonate metabolism gene phnH, dissimilatory nitrate reductase subunit napA) to >2.7 × 10(9) transcripts per liter (ammonia transporter amt and ammonia monooxygenase subunit amoC). Half of the categories for which expression was detected, however, had too few copy numbers for robust statistical resolution, as would be required for comparative (experimental or time-series) expression studies. By representing whole community gene abundance and expression in absolute units (per volume or mass of environment), 'omics' data can be better leveraged to improve understanding of microbially mediated processes in the ocean.
Figures
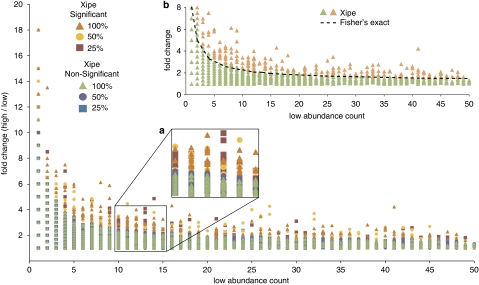
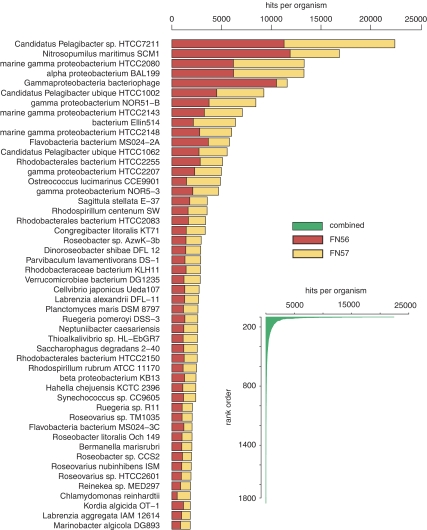
References
-
- Azam F, Hodson RE. Size distribution and activity of marine microheterotrophs. Limnol Oceanogr. 1977;22:492–501.
-
- Bürgmann H, Howard EC, Ye WY, Sun F, Sun SL, Napierala S, et al. Transcriptional response of Silicibacter pomeroyi DSS-3 to dimethylsulfoniopropionate (DMSP) Environ Microbiol. 2007;9:2742–2755. - PubMed
-
- Campbell BJ, Waidner LA, Cottrell MT, Kirchman DL. Abundant proteorhodopsin genes in the North Atlantic Ocean. Environ Microbiol. 2008;10:99–109. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
