

The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells
- PMID: 20844577
- PMCID: PMC2936548
- DOI: 10.1371/journal.ppat.1001099
The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells
Abstract
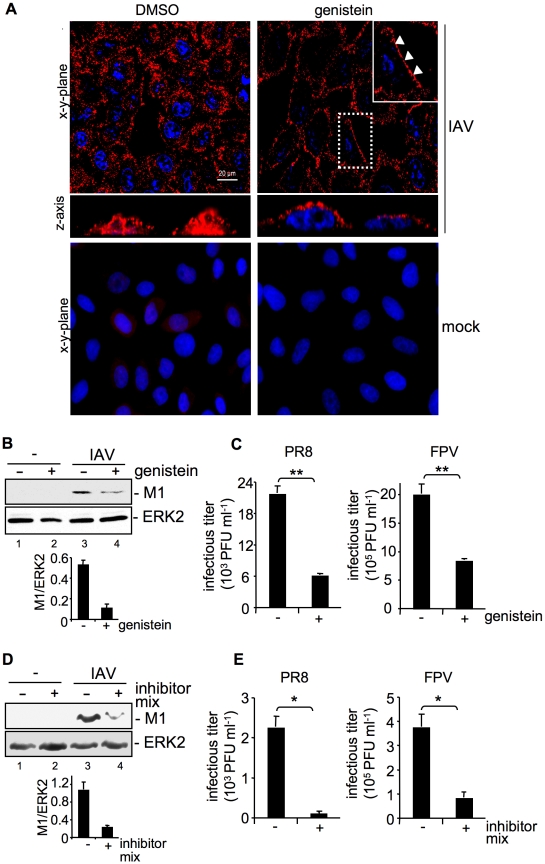
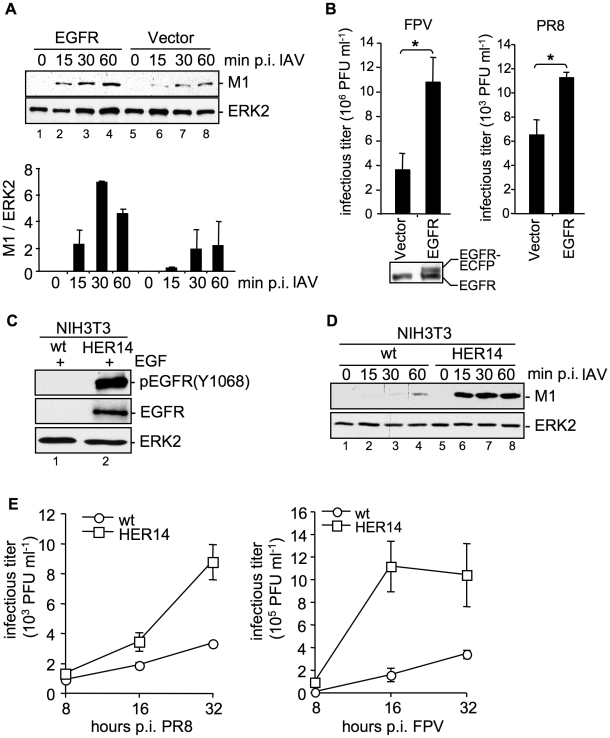
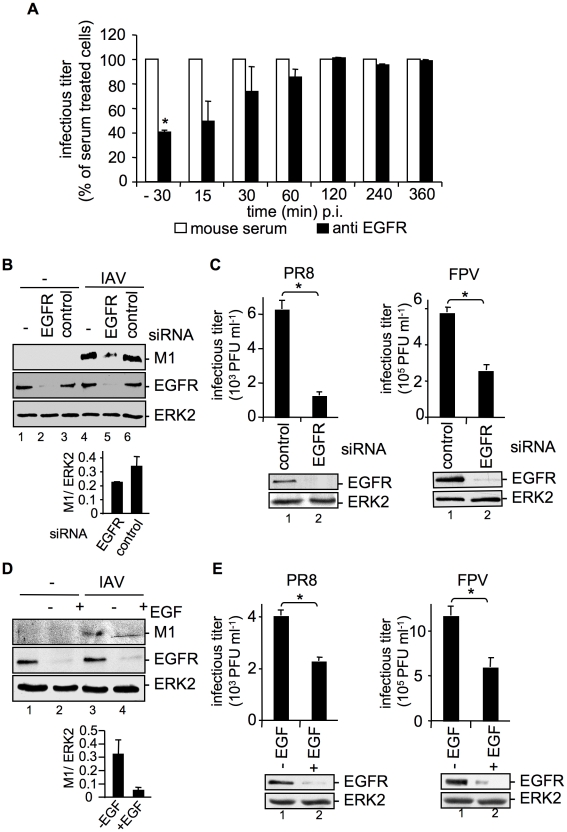
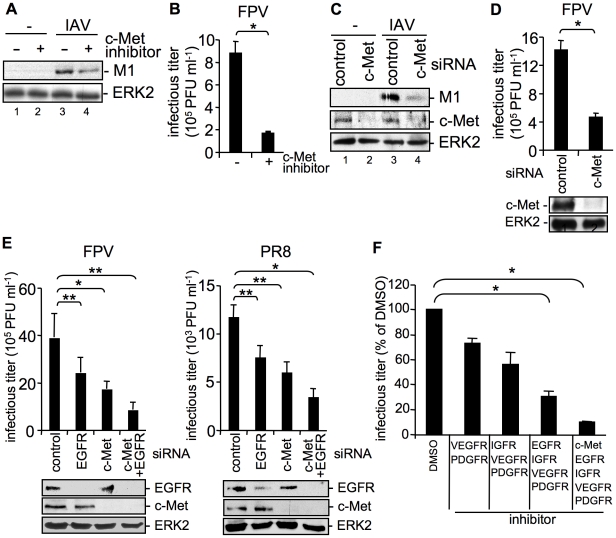
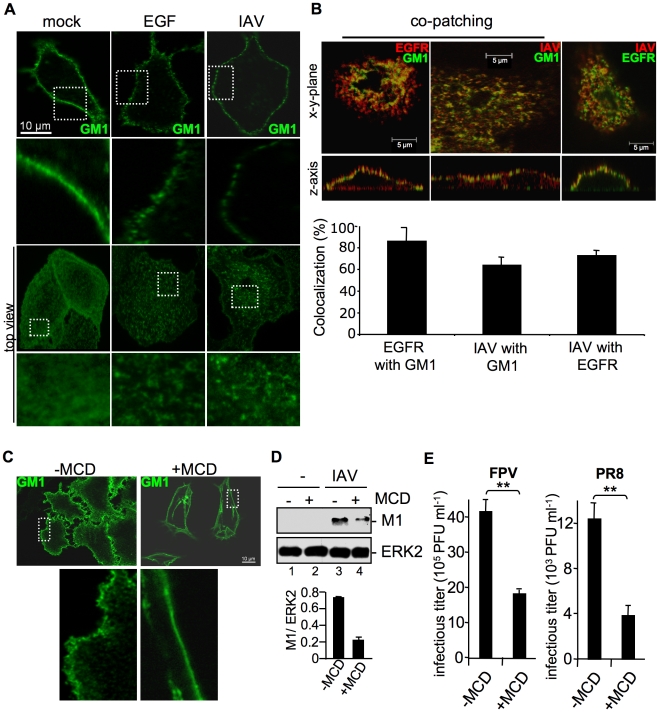
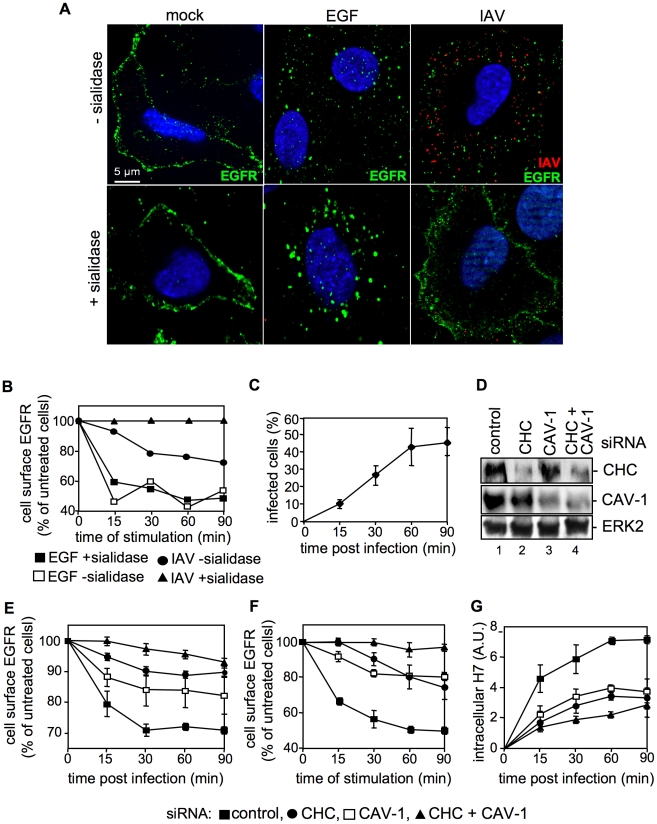
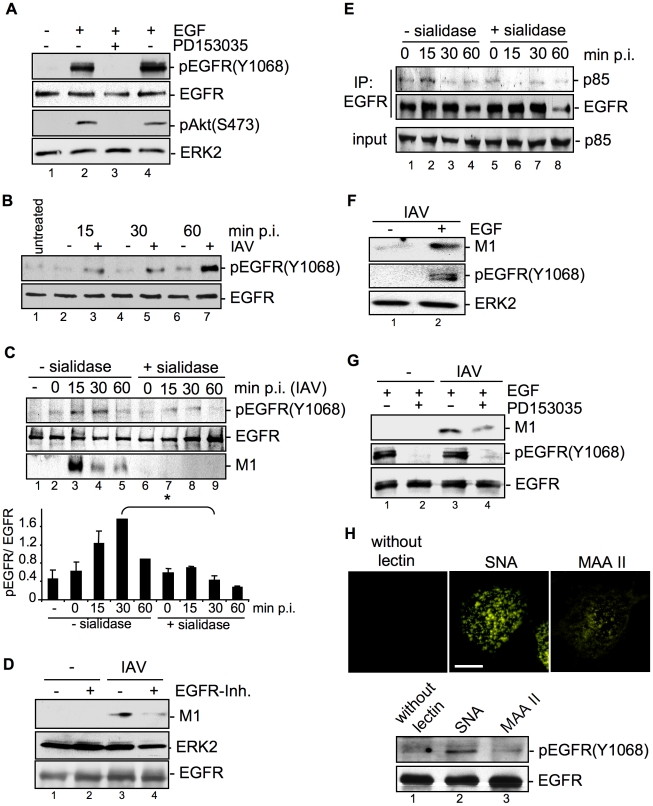
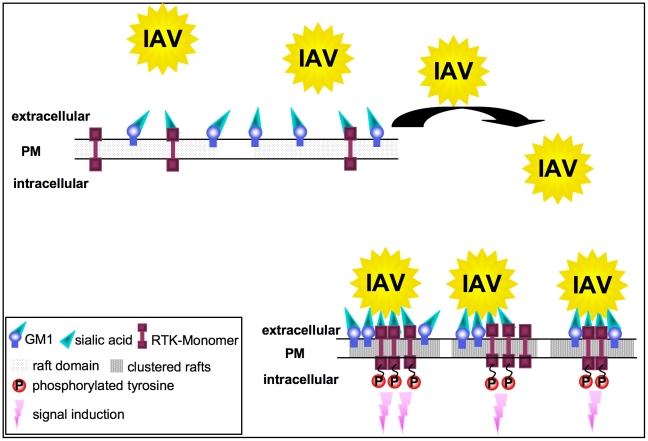
Influenza A viruses (IAV) bind to sialic-acids at cellular surfaces and enter cells by using endocytotic routes. There is evidence that this process does not occur constitutively but requires induction of specific cellular signals, including activation of PI3K that promotes virus internalization. This implies engagement of cellular signaling receptors during viral entry. Here, we present first indications for an interplay of IAV with receptor tyrosine kinases (RTKs). As representative RTK family-members the epidermal growth factor receptor (EGFR) and the c-Met receptor were studied. Modulation of expression or activity of both RTKs resulted in altered uptake of IAV, showing that these receptors transmit entry relevant signals upon virus binding. More detailed studies on EGFR function revealed that virus binding lead to clustering of lipid-rafts, suggesting that multivalent binding of IAV to cells induces a signaling platform leading to activation of EGFR and other RTKs that in turn facilitates IAV uptake.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Pleschka S, Wolff T, Ehrhardt C, Hobom G, Planz O, et al. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat Cell Biol. 2001;3:301–305. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
