

Dendritic cell-specific antigen delivery by coronavirus vaccine vectors induces long-lasting protective antiviral and antitumor immunity
- PMID: 20844609
- PMCID: PMC2939679
- DOI: 10.1128/mBio.00171-10
Dendritic cell-specific antigen delivery by coronavirus vaccine vectors induces long-lasting protective antiviral and antitumor immunity
Abstract
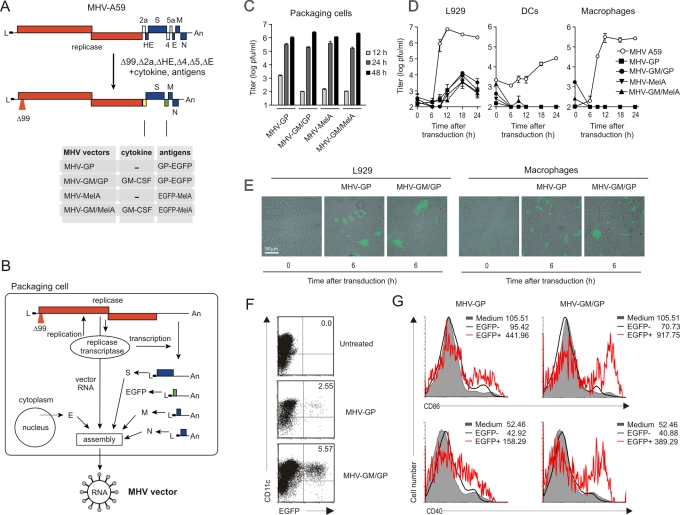
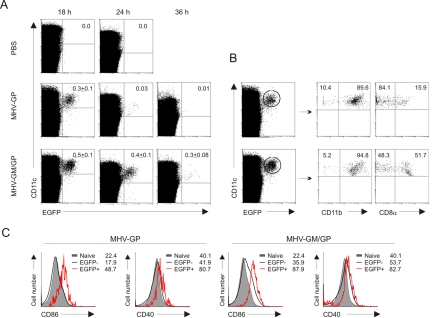
Efficient vaccination against infectious agents and tumors depends on specific antigen targeting to dendritic cells (DCs). We report here that biosafe coronavirus-based vaccine vectors facilitate delivery of multiple antigens and immunostimulatory cytokines to professional antigen-presenting cells in vitro and in vivo. Vaccine vectors based on heavily attenuated murine coronavirus genomes were generated to express epitopes from the lymphocytic choriomeningitis virus glycoprotein, or human Melan-A, in combination with the immunostimulatory cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF). These vectors selectively targeted DCs in vitro and in vivo resulting in vector-mediated antigen expression and efficient maturation of DCs. Single application of only low vector doses elicited strong and long-lasting cytotoxic T-cell responses, providing protective antiviral and antitumor immunity. Furthermore, human DCs transduced with Melan-A-recombinant human coronavirus 229E efficiently activated tumor-specific CD8(+) T cells. Taken together, this novel vaccine platform is well suited to deliver antigens and immunostimulatory cytokines to DCs and to initiate and maintain protective immunity.
Figures
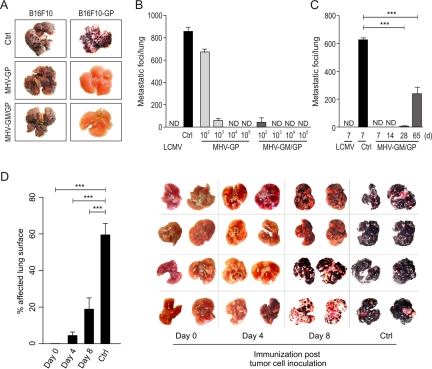
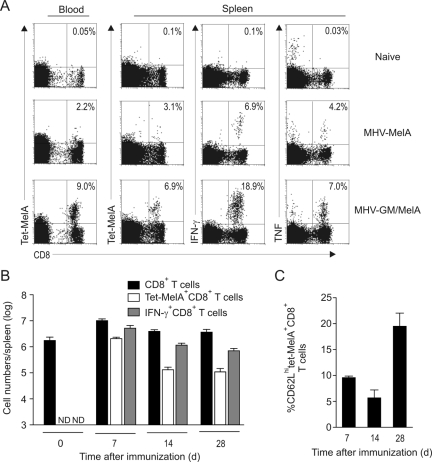
References
-
- McMichael A. J. 2006. HIV vaccines. Annu. Rev. Immunol. 24:227–255 - PubMed
-
- Strickland G. T., El Kamary S. S., Klenerman P., Nicosia A. 2008. Hepatitis C vaccine: supply and demand. Lancet Infect. Dis. 8:379–386 - PubMed
-
- Melief C. J. 2008. Cancer immunotherapy by dendritic cells. Immunity 29:372–383 - PubMed
-
- Appay V., Douek D. C., Price D. A. 2008. CD8+ T cell efficacy in vaccination and disease. Nat. Med. 14:623–628 - PubMed
-
- Tacken P. J., de Vries I. J., Torensma R., Figdor C. G. 2007. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat. Rev. Immunol. 7:790–802 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
