

Evidence for an alternative glycolytic pathway in rapidly proliferating cells
- PMID: 20847263
- PMCID: PMC3030121
- DOI: 10.1126/science.1188015
Evidence for an alternative glycolytic pathway in rapidly proliferating cells
Abstract
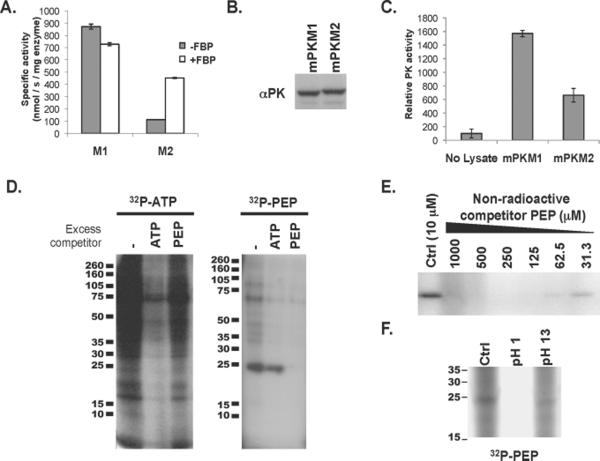
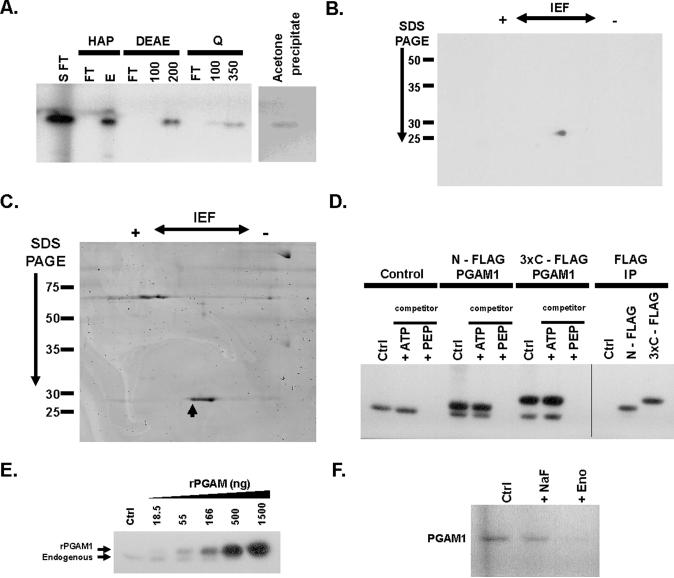
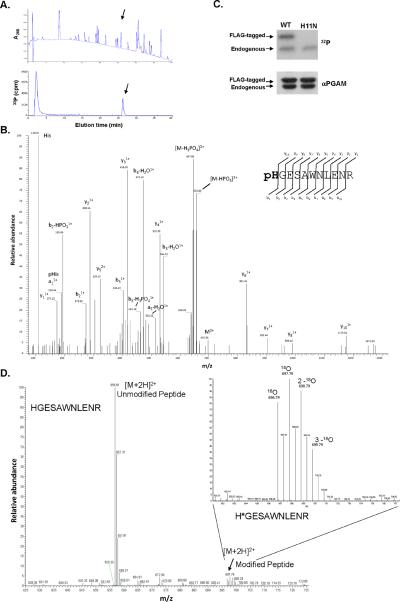
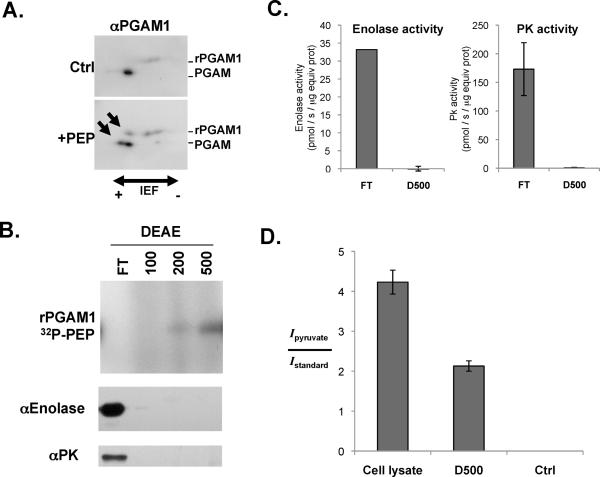
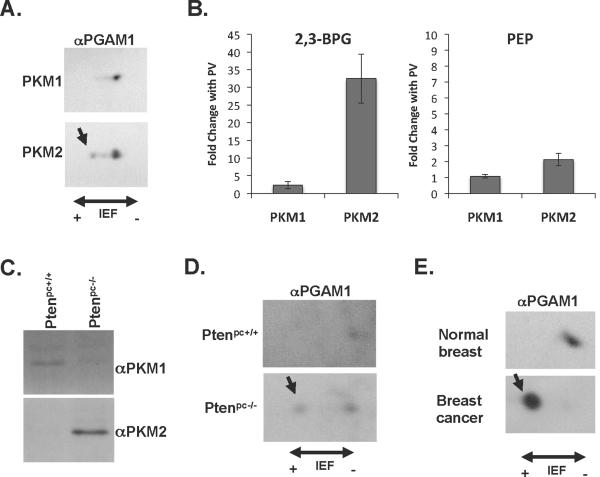
Proliferating cells, including cancer cells, require altered metabolism to efficiently incorporate nutrients such as glucose into biomass. The M2 isoform of pyruvate kinase (PKM2) promotes the metabolism of glucose by aerobic glycolysis and contributes to anabolic metabolism. Paradoxically, decreased pyruvate kinase enzyme activity accompanies the expression of PKM2 in rapidly dividing cancer cells and tissues. We demonstrate that phosphoenolpyruvate (PEP), the substrate for pyruvate kinase in cells, can act as a phosphate donor in mammalian cells because PEP participates in the phosphorylation of the glycolytic enzyme phosphoglycerate mutase (PGAM1) in PKM2-expressing cells. We used mass spectrometry to show that the phosphate from PEP is transferred to the catalytic histidine (His11) on human PGAM1. This reaction occurred at physiological concentrations of PEP and produced pyruvate in the absence of PKM2 activity. The presence of histidine-phosphorylated PGAM1 correlated with the expression of PKM2 in cancer cell lines and tumor tissues. Thus, decreased pyruvate kinase activity in PKM2-expressing cells allows PEP-dependent histidine phosphorylation of PGAM1 and may provide an alternate glycolytic pathway that decouples adenosine triphosphate production from PEP-mediated phosphotransfer, allowing for the high rate of glycolysis to support the anabolic metabolism observed in many proliferating cells.
Figures
Comment in
-
Metabolism: Less is sometimes more.Nat Rev Cancer. 2010 Nov;10(11):740. doi: 10.1038/nrc2954. Nat Rev Cancer. 2010. PMID: 21080591 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- R21/R33 DK070299/DK/NIDDK NIH HHS/United States
- P01GM047467/GM/NIGMS NIH HHS/United States
- T32 CA009172/CA/NCI NIH HHS/United States
- P01 GM047467/GM/NIGMS NIH HHS/United States
- R33 DK070299/DK/NIDDK NIH HHS/United States
- 1K08CA136983/CA/NCI NIH HHS/United States
- 1P01CA120964-01A/CA/NCI NIH HHS/United States
- P01 CA089021/CA/NCI NIH HHS/United States
- T32 CA009361/CA/NCI NIH HHS/United States
- R01-GM56302/GM/NIGMS NIH HHS/United States
- 5 T32 CA009361-28/CA/NCI NIH HHS/United States
- P01 CA120964/CA/NCI NIH HHS/United States
- R21 CA128620/CA/NCI NIH HHS/United States
- F32 CA009361/CA/NCI NIH HHS/United States
- R01 AI078063/AI/NIAID NIH HHS/United States
- P01CA089021/CA/NCI NIH HHS/United States
- K08 CA136983/CA/NCI NIH HHS/United States
- 5P30CA006516-43/CA/NCI NIH HHS/United States
- P30 CA006516/CA/NCI NIH HHS/United States
- R21 DK070299/DK/NIDDK NIH HHS/United States
- R01 GM056203/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
