

The archaeal topoisomerase reverse gyrase is a helix-destabilizing protein that unwinds four-way DNA junctions
- PMID: 20851892
- PMCID: PMC2978581
- DOI: 10.1074/jbc.M110.169029
The archaeal topoisomerase reverse gyrase is a helix-destabilizing protein that unwinds four-way DNA junctions
Abstract
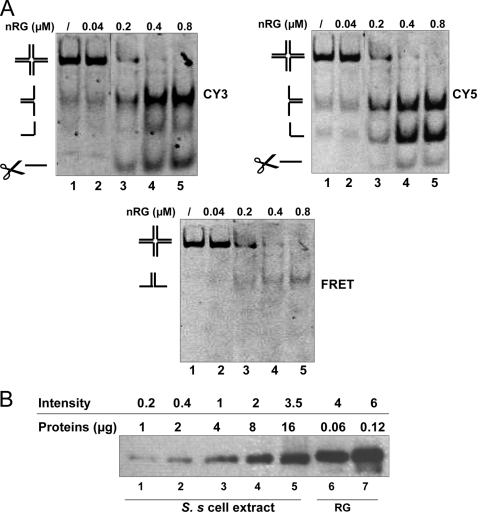
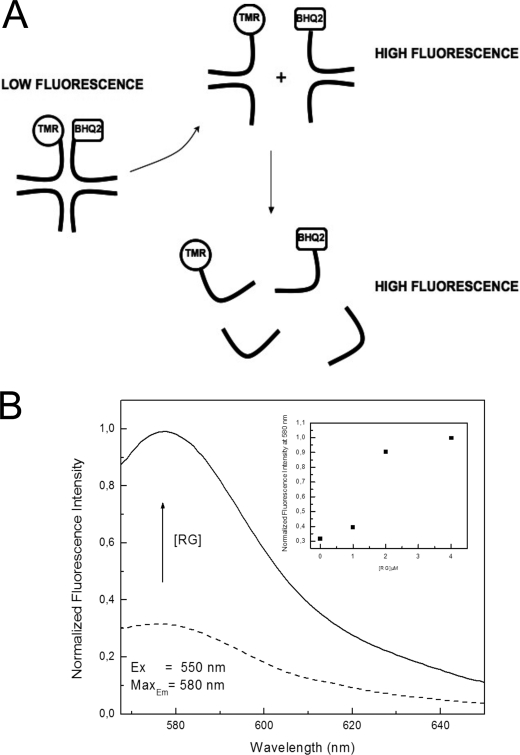
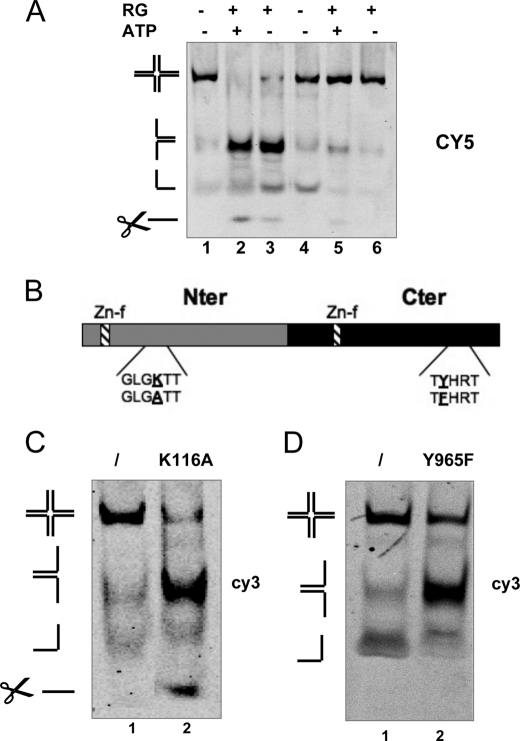
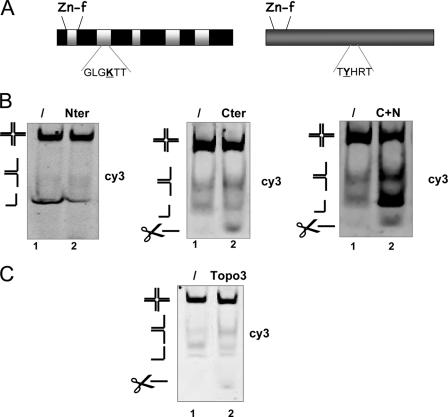
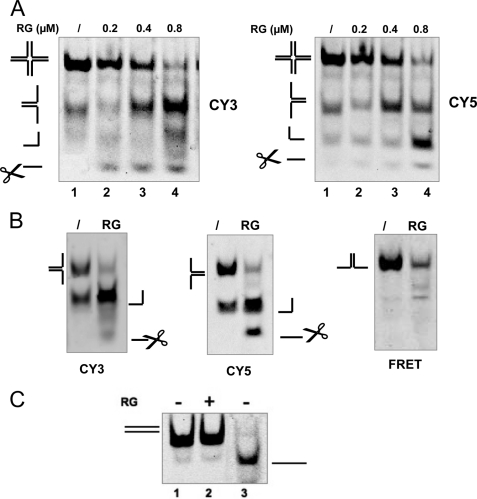
Four-way junctions are non-B DNA structures that originate as intermediates of recombination and repair (Holliday junctions) or from the intrastrand annealing of palindromic sequences (cruciforms). These structures have important functional roles but may also severely interfere with DNA replication and other genetic processes; therefore, they are targeted by regulatory and architectural proteins, and dedicated pathways exist for their removal. Although it is well known that resolution of Holliday junctions occurs either by recombinases or by specialized helicases, less is known on the mechanisms dealing with secondary structures in nucleic acids. Reverse gyrase is a DNA topoisomerase, specific to microorganisms living at high temperatures, which comprises a type IA topoisomerase fused to an SF2 helicase-like module and catalyzes ATP hydrolysis-dependent DNA positive supercoiling. Reverse gyrase is likely involved in regulation of DNA structure and stability and might also participate in the cell response to DNA damage. By applying FRET technology to multiplex fluorophore gel imaging, we show here that reverse gyrase induces unwinding of synthetic four-way junctions as well as forked DNA substrates, following a mechanism independent of both the ATPase and the strand-cutting activity of the enzyme. The reaction requires high temperature and saturating protein concentrations. Our results suggest that reverse gyrase works like an ATP-independent helix-destabilizing protein specific for branched DNA structures. The results are discussed in light of reverse gyrase function and their general relevance for protein-mediated unwinding of complex DNA structures.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
