

Deletion of Pten in pancreatic ß-cells protects against deficient ß-cell mass and function in mouse models of type 2 diabetes
- PMID: 20852026
- PMCID: PMC2992773
- DOI: 10.2337/db09-1805
Deletion of Pten in pancreatic ß-cells protects against deficient ß-cell mass and function in mouse models of type 2 diabetes
Abstract
Objective: Type 2 diabetes is characterized by diminished pancreatic β-cell mass and function. Insulin signaling within the β-cells has been shown to play a critical role in maintaining the essential function of the β-cells. Under basal conditions, enhanced insulin-PI3K signaling via deletion of phosphatase with tensin homology (PTEN), a negative regulator of this pathway, leads to increased β-cell mass and function. In this study, we investigated the effects of prolonged β-cell-specific PTEN deletion in models of type 2 diabetes.
Research design and methods: Two models of type 2 diabetes were employed: a high-fat diet (HFD) model and a db/db model that harbors a global leptin-signaling defect. A Cre-loxP system driven by the rat insulin promoter (RIP) was employed to obtain mice with β-cell-specific PTEN deletion (RIPcre(+) Pten(fl/fl)).
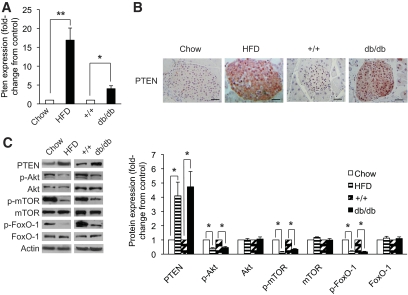
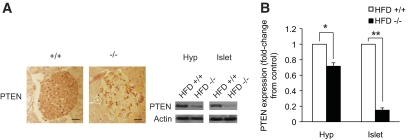
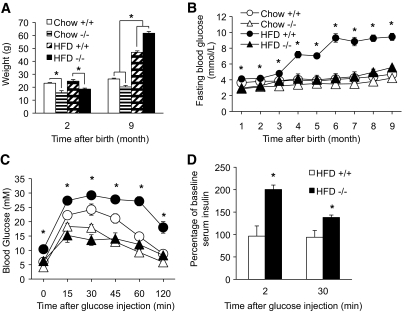
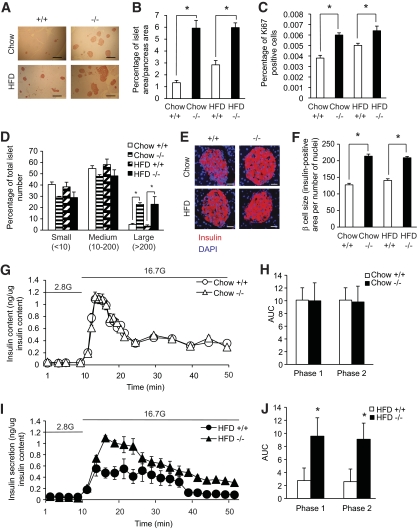
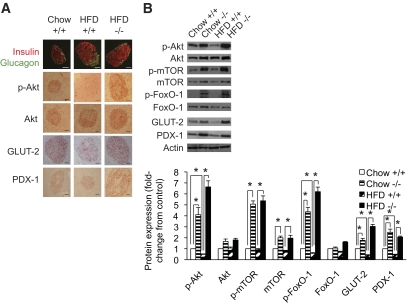
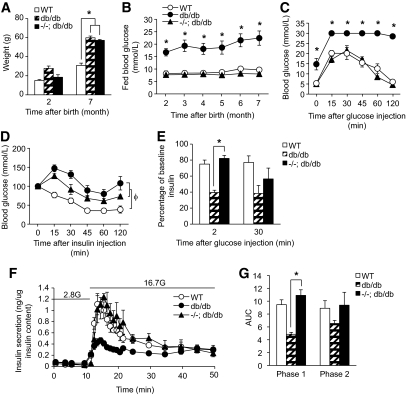
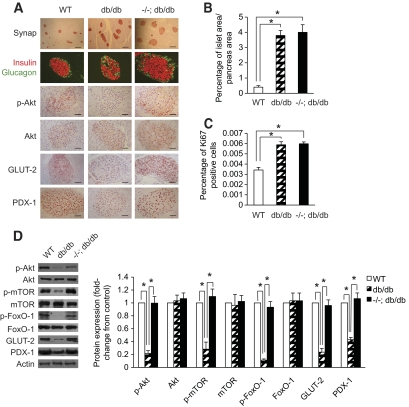
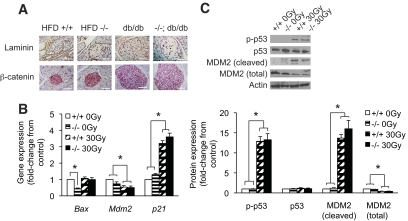
Results: PTEN expression in islets was upregulated in both models of type 2 diabetes. RIPcre(+) Pten(fl/fl) mice were completely protected against diabetes in both models of type 2 diabetes. The islets of RIPcre(+) Pten(fl/fl) mice already exhibited increased β-cell mass under basal conditions, and there was no further increase under diabetic conditions. Their β-cell function and islet PI3K signaling remained intact, in contrast to HFD-fed wild-type and db/db islets that exhibited diminished β-cell function and attenuated PI3K signaling. These protective effects in β-cells occurred in the absence of compromised response to DNA-damaging stimuli.
Conclusions: PTEN exerts a critical negative effect on both β-cell mass and function. Thus PTEN inhibition in β-cells can be a novel therapeutic intervention to prevent the decline of β-cell mass and function in type 2 diabetes.
Figures
Similar articles
-
Overexpression of microRNA-26a protects against deficient β-cell function via targeting phosphatase with tensin homology in mouse models of type 2 diabetes.Biochem Biophys Res Commun. 2018 Jan 1;495(1):1312-1316. doi: 10.1016/j.bbrc.2017.11.170. Epub 2017 Dec 2. Biochem Biophys Res Commun. 2018. PMID: 29191656
-
Selective disruption of Tcf7l2 in the pancreatic β cell impairs secretory function and lowers β cell mass.Hum Mol Genet. 2015 Mar 1;24(5):1390-9. doi: 10.1093/hmg/ddu553. Epub 2014 Oct 29. Hum Mol Genet. 2015. PMID: 25355422 Free PMC article.
-
Metallothionein 1 negatively regulates glucose-stimulated insulin secretion and is differentially expressed in conditions of beta cell compensation and failure in mice and humans.Diabetologia. 2019 Dec;62(12):2273-2286. doi: 10.1007/s00125-019-05008-3. Epub 2019 Oct 17. Diabetologia. 2019. PMID: 31624901
-
Role of VEGF-A in pancreatic beta cells.Endocr J. 2010;57(3):185-91. doi: 10.1507/endocrj.k09e-035. Epub 2010 Feb 24. Endocr J. 2010. PMID: 20179357 Review.
-
Akt and PTEN: beta-cell mass and pancreas plasticity.Trends Endocrinol Metab. 2009 Jul;20(5):243-51. doi: 10.1016/j.tem.2009.03.002. Epub 2009 Jun 21. Trends Endocrinol Metab. 2009. PMID: 19541499 Free PMC article. Review.
Cited by
-
A new role for the brain in metabolic control.Nat Med. 2014 May;20(5):472-3. doi: 10.1038/nm.3556. Nat Med. 2014. PMID: 24804753 No abstract available.
-
Tet2 Controls the Responses of β cells to Inflammation in Autoimmune Diabetes.Nat Commun. 2021 Aug 20;12(1):5074. doi: 10.1038/s41467-021-25367-z. Nat Commun. 2021. PMID: 34417463 Free PMC article.
-
β-Cell Insulin Resistance Plays a Causal Role in Fat-Induced β-Cell Dysfunction In Vitro and In Vivo.Endocrinology. 2024 Mar 29;165(5):bqae044. doi: 10.1210/endocr/bqae044. Endocrinology. 2024. PMID: 38578954 Free PMC article.
-
Good news for the ageing beta cell.Diabetologia. 2014 Feb;57(2):265-9. doi: 10.1007/s00125-013-3114-7. Epub 2013 Nov 21. Diabetologia. 2014. PMID: 24257895 Free PMC article. No abstract available.
-
Ras inhibition induces insulin sensitivity and glucose uptake.PLoS One. 2011;6(6):e21712. doi: 10.1371/journal.pone.0021712. Epub 2011 Jun 29. PLoS One. 2011. PMID: 21738773 Free PMC article.
References
-
- Marchetti P, Del Guerra S, Marselli L, Lupi R, Masini M, Pollera M, Bugliani M, Boggi U, Vistoli F, Mosca F, Del Prato S: Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J Clin Endocrinol Metab 2004;89:5535–5541 - PubMed
-
- Mitrakou A, Kelley D, Mokan M, Veneman T, Pangburn T, Reilly J, Gerich J: Role of reduced suppression of glucose production and diminished early insulin release in impaired glucose tolerance. N Engl J Med 1992;326:22–29 - PubMed
-
- Porte D, Jr, Kahn SE: β-cell dysfunction and failure in type 2 diabetes: potential mechanisms. Diabetes 2001;1(Suppl.):S160–S163 - PubMed
-
- Kahn SE: β cell failure: causes and consequences. Int J Clin Pract 2001;September:13–18 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
