

A novel cerebello-ocular syndrome with abnormal glycosylation due to abnormalities in dolichol metabolism
- PMID: 20852264
- PMCID: PMC6276930
- DOI: 10.1093/brain/awq261
A novel cerebello-ocular syndrome with abnormal glycosylation due to abnormalities in dolichol metabolism
Abstract
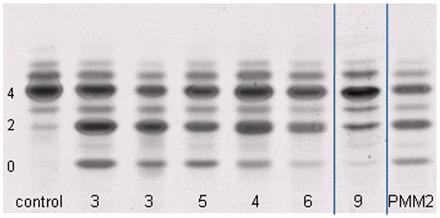
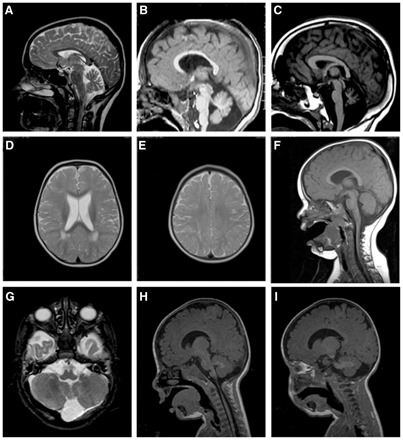
Cerebellar hypoplasia and slowly progressive ophthalmological symptoms are common features in patients with congenital disorders of glycosylation type I. In a group of patients with congenital disorders of glycosylation type I with unknown aetiology, we have previously described a distinct phenotype with severe, early visual impairment and variable eye malformations, including optic nerve hypoplasia, retinal coloboma, congenital cataract and glaucoma. Some of the symptoms overlapped with the phenotype in other congenital disorders of glycosylation type I subtypes, such as vermis hypoplasia, anaemia, ichtyosiform dermatitis, liver dysfunction and coagulation abnormalities. We recently identified pathogenic mutations in the SRD5A3 gene, encoding steroid 5α-reductase type 3, in a group of patients who presented with this particular phenotype and a common metabolic pattern. Here, we report on the clinical, genetic and metabolic features of 12 patients from nine families with cerebellar ataxia and congenital eye malformations diagnosed with SRD5A3-congenital disorders of glycosylation due to steroid 5α-reductase type 3 defect. This enzyme is necessary for the reduction of polyprenol to dolichol, the lipid anchor for N-glycosylation in the endoplasmic reticulum. Dolichol synthesis is an essential metabolic step in protein glycosylation. The current defect leads to a severely abnormal glycosylation state already in the early phase of the N-glycan biosynthesis pathway in the endoplasmic reticulum. We detected high expression of SRD5A3 in foetal brain tissue, especially in the cerebellum, consistent with the finding of the congenital cerebellar malformations. Based on the overlapping clinical, biochemical and genetic data in this large group of patients with congenital disorders of glycosylation, we define a novel syndrome of cerebellar ataxia associated with congenital eye malformations due to a defect in dolichol metabolism.
Figures


References
-
- Al-Gazali L, Hertecant J, Algawi K, El Teraifi H, Dattani M. A new autosomal recessive syndrome of ocular colobomas, ichthyosis, brain malformations and endocrine abnormalities in an inbred Emirati family. Am J Med Genet A. 2008;146:813–9. - PubMed
-
- Assmann B, Hackler R, Peters V, Schaefer JR, Arndt T, Mayatepek E, et al. A new subtype of a congenital disorder of glycosylation (CDG) with mild clinical manifestations. Neuropediatrics. 2001;32:313–8. - PubMed
-
- Babovic-Vuksanovic D, O'Brien JF. Laboratory diagnosis of congenital disorders of glycosylation type I by analysis of transferrin glycoforms. Mol Diagn Ther. 2007;11:303–11. - PubMed
-
- Bickel T, Lehle L, Schwarz M, Aebi M, Jakob CA. Biosynthesis of lipid-linked oligosaccharides in Saccharomyces cerevisiae: ALG13p and ALG14p form a complex required for the formation of GlcNAc2-PP-Dol. J Biol Chem. 2005;280:34500–6. - PubMed
-
- Brownstein MJ, Carpten JD, Smith JR. Modulation of non-templated nucleotide addition by Taq DNA polymerase: primer modifications that facilitate genotyping. Biotechniques. 1996;20:1004–10. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
