

PPARγ-induced cardiolipotoxicity in mice is ameliorated by PPARα deficiency despite increases in fatty acid oxidation
- PMID: 20852389
- PMCID: PMC2947216
- DOI: 10.1172/JCI40905
PPARγ-induced cardiolipotoxicity in mice is ameliorated by PPARα deficiency despite increases in fatty acid oxidation
Erratum in
- J Clin Invest. 2010 Dec 1;120(12):4583
Abstract
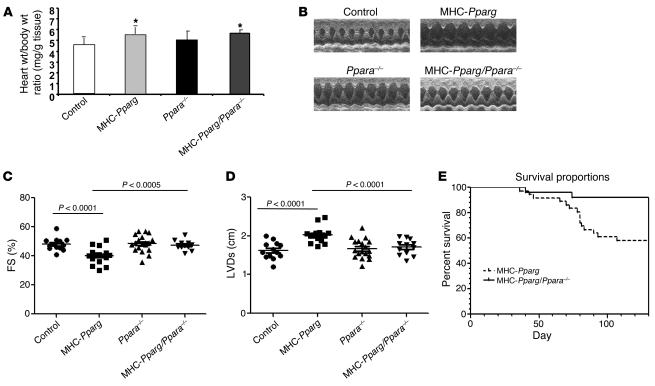
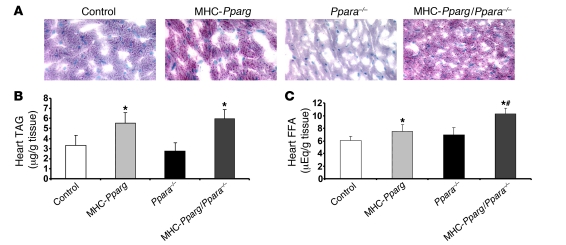
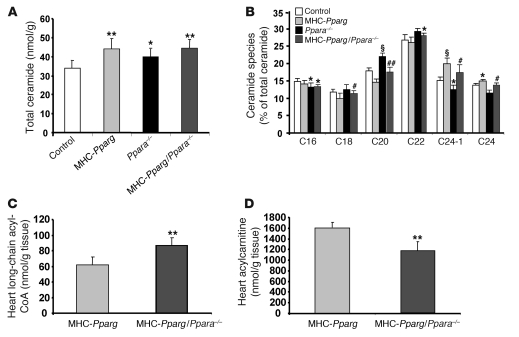
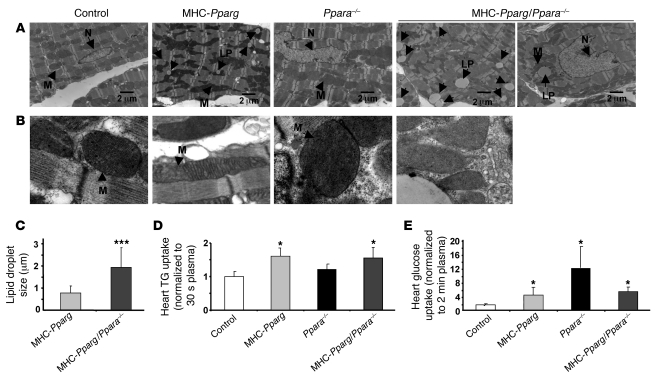
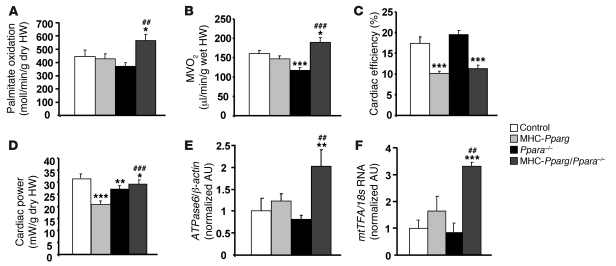
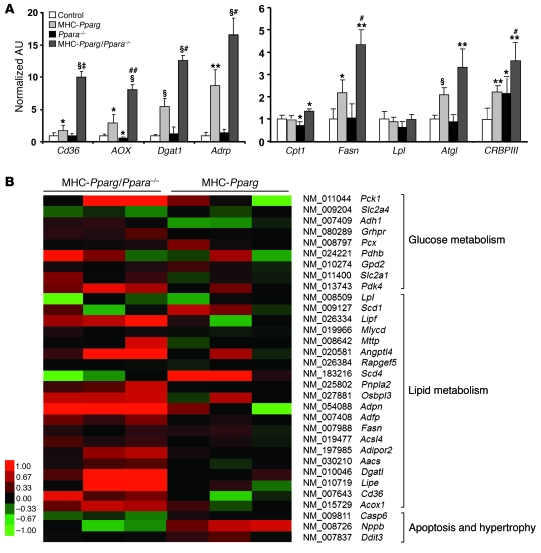
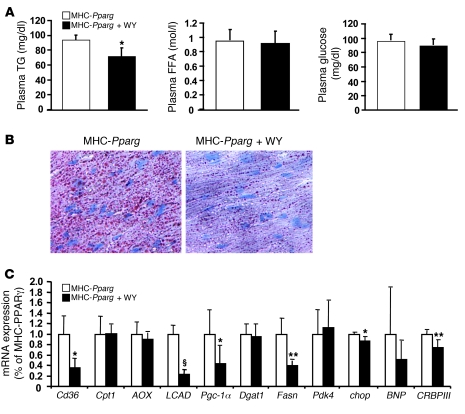
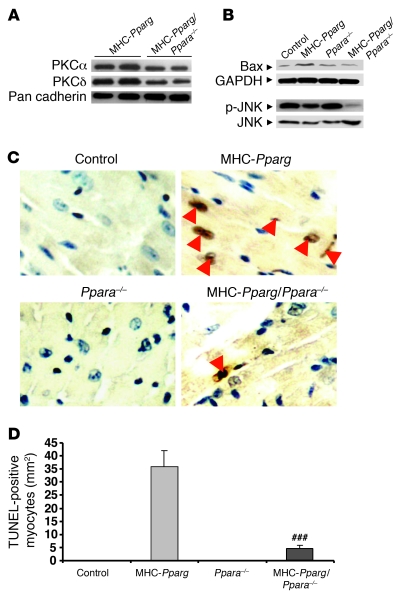
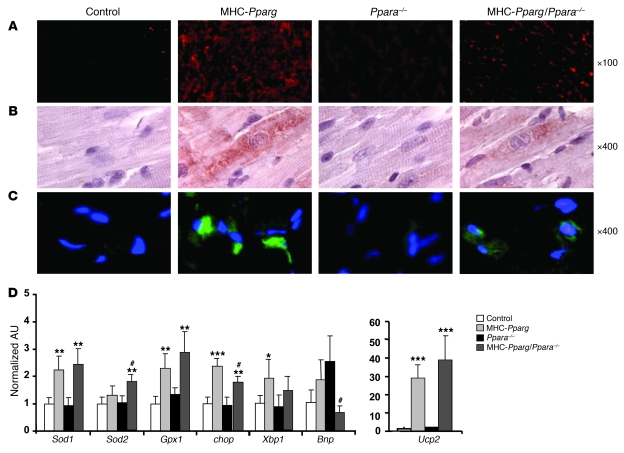
Excess lipid accumulation in the heart is associated with decreased cardiac function in humans and in animal models. The reasons are unclear, but this is generally believed to result from either toxic effects of intracellular lipids or excessive fatty acid oxidation (FAO). PPARγ expression is increased in the hearts of humans with metabolic syndrome, and use of PPARγ agonists is associated with heart failure. Here, mice with dilated cardiomyopathy due to cardiomyocyte PPARγ overexpression were crossed with PPARα-deficient mice. Surprisingly, this cross led to enhanced expression of several PPAR-regulated genes that mediate fatty acid (FA) uptake/oxidation and triacylglycerol (TAG) synthesis. Although FA oxidation and TAG droplet size were increased, heart function was preserved and survival improved. There was no marked decrease in cardiac levels of triglyceride or the potentially toxic lipids diacylglycerol (DAG) and ceramide. However, long-chain FA coenzyme A (LCCoA) levels were increased, and acylcarnitine content was decreased. Activation of PKCα and PKCδ, apoptosis, ROS levels, and evidence of endoplasmic reticulum stress were also reduced. Thus, partitioning of lipid to storage and oxidation can reverse cardiolipotoxicity despite increased DAG and ceramide levels, suggesting a role for other toxic intermediates such as acylcarnitines in the toxic effects of lipid accumulation in the heart.
Figures
Comment in
-
Lipotoxicity and the development of heart failure: moving from mouse to man.Cell Metab. 2010 Dec 1;12(6):555-6. doi: 10.1016/j.cmet.2010.11.016. Cell Metab. 2010. PMID: 21109186 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
