

Quantitative comparison of genome-wide DNA methylation mapping technologies
- PMID: 20852634
- PMCID: PMC3066564
- DOI: 10.1038/nbt.1681
Quantitative comparison of genome-wide DNA methylation mapping technologies
Abstract
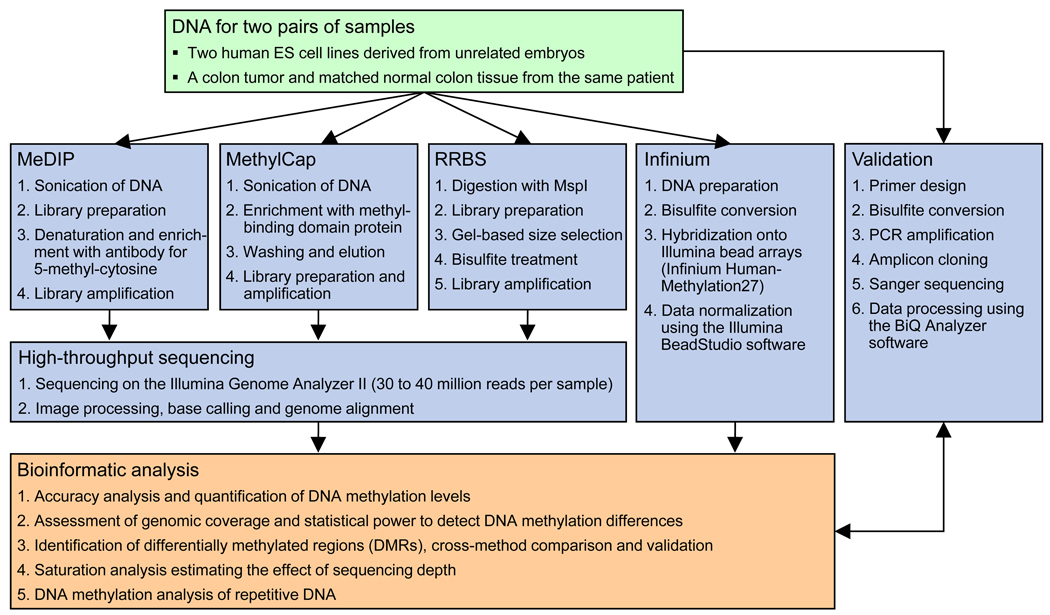
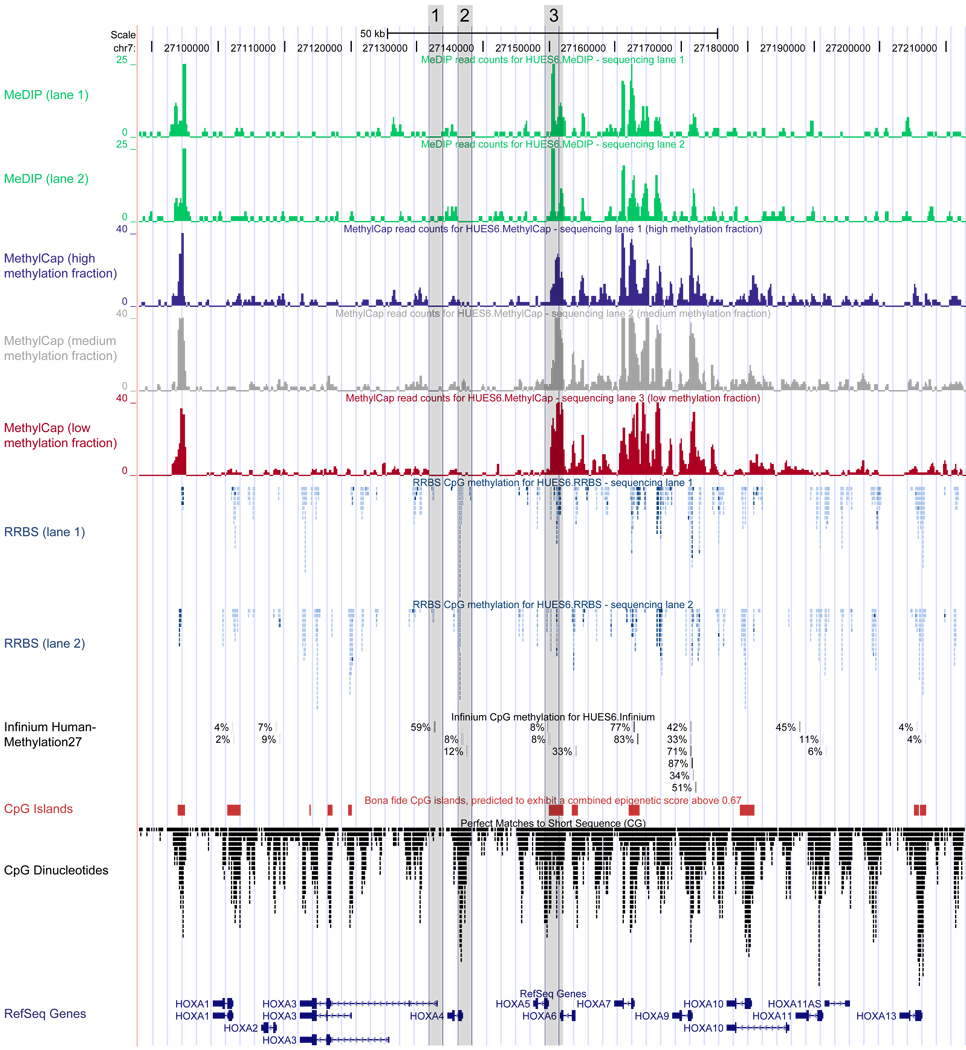
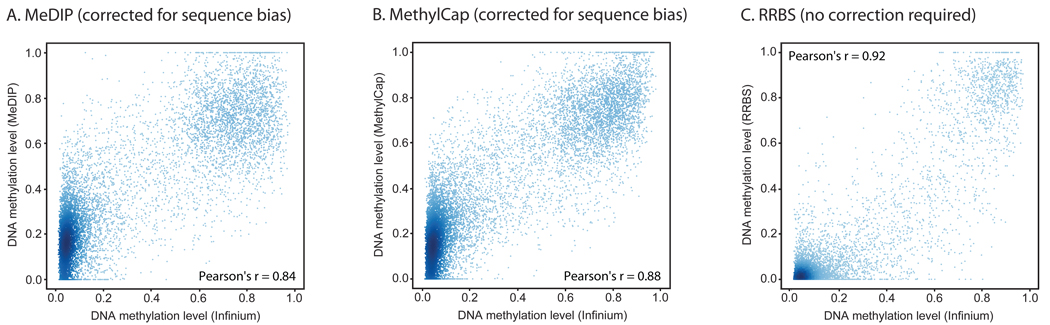
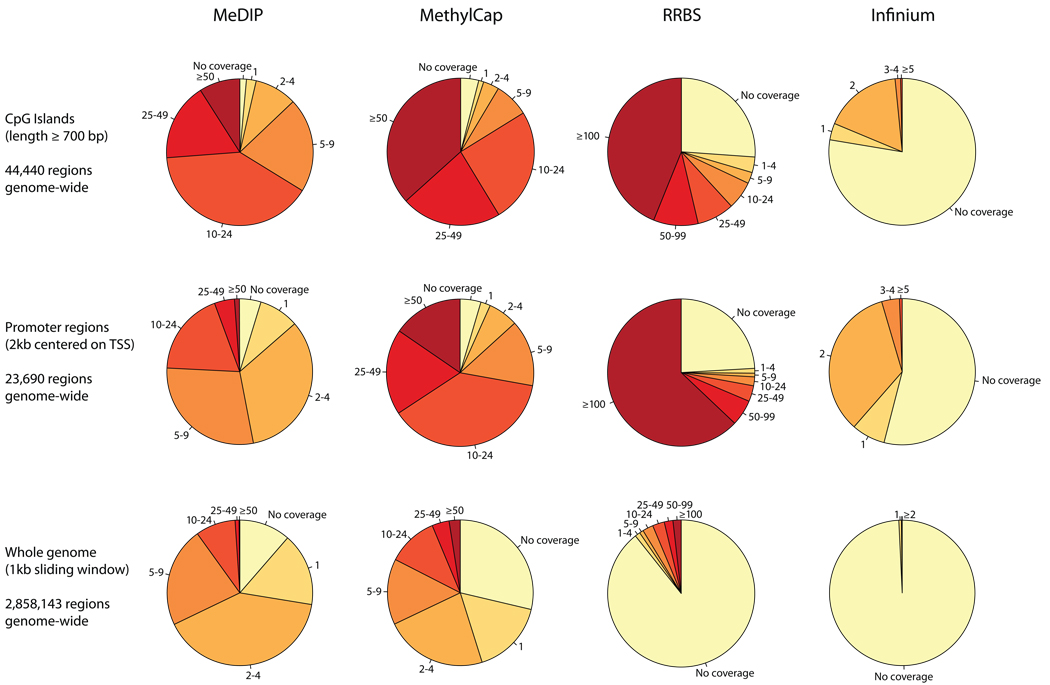
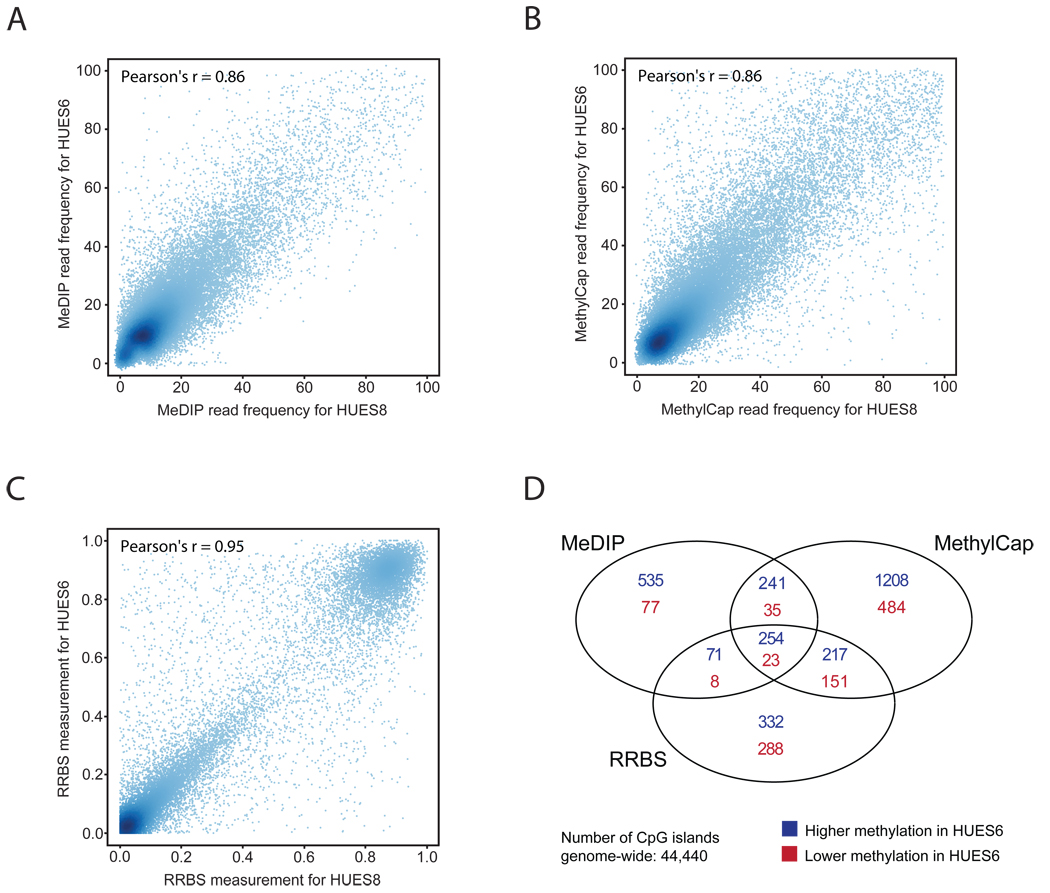
DNA methylation plays a key role in regulating eukaryotic gene expression. Although mitotically heritable and stable over time, patterns of DNA methylation frequently change in response to cell differentiation, disease and environmental influences. Several methods have been developed to map DNA methylation on a genomic scale. Here, we benchmark four of these approaches by analyzing two human embryonic stem cell lines derived from genetically unrelated embryos and a matched pair of colon tumor and adjacent normal colon tissue obtained from the same donor. Our analysis reveals that methylated DNA immunoprecipitation sequencing (MeDIP-seq), methylated DNA capture by affinity purification (MethylCap-seq), reduced representation bisulfite sequencing (RRBS) and the Infinium HumanMethylation27 assay all produce accurate DNA methylation data. However, these methods differ in their ability to detect differentially methylated regions between pairs of samples. We highlight strengths and weaknesses of the four methods and give practical recommendations for the design of epigenomic case-control studies.
Figures
Comment in
-
Taking the measure of the methylome.Nat Biotechnol. 2010 Oct;28(10):1026-8. doi: 10.1038/nbt1010-1026. Nat Biotechnol. 2010. PMID: 20944589
References
-
- Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6(2):107–116. - PubMed
-
- Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148–1159. - PubMed
-
- Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4(2):143–153. - PubMed
-
- Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4(12):988–993. - PubMed
-
- Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21(2):163–167. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
