

Ligand-accelerated C-H activation reactions: evidence for a switch of mechanism
- PMID: 20853838
- PMCID: PMC2952432
- DOI: 10.1021/ja105044s
Ligand-accelerated C-H activation reactions: evidence for a switch of mechanism
Abstract
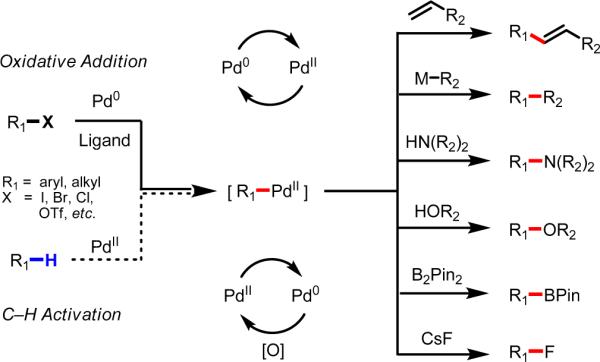
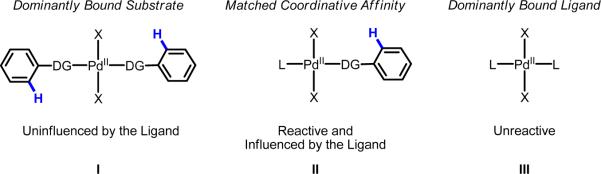
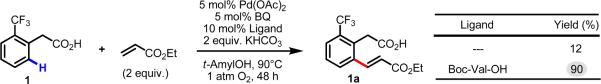
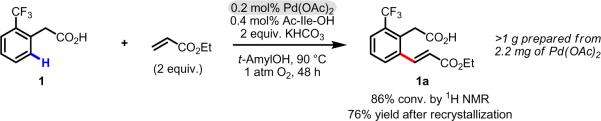
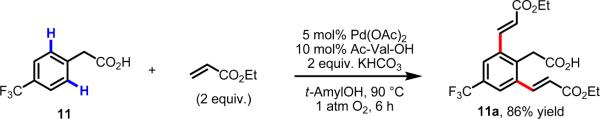
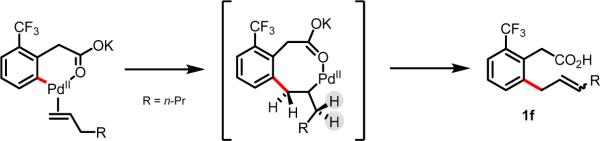
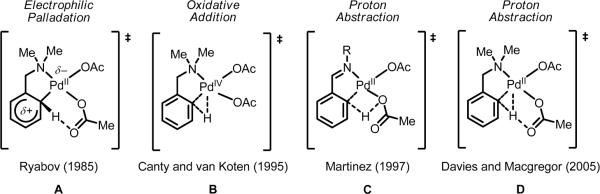
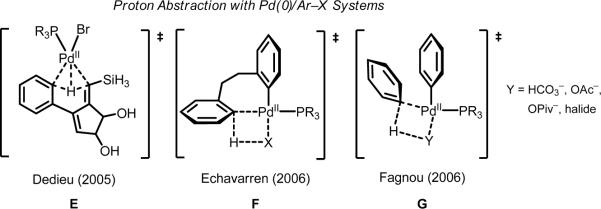
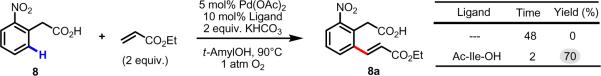
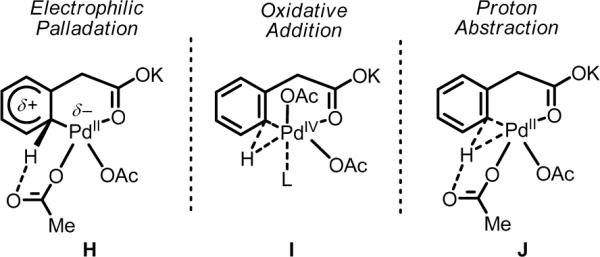
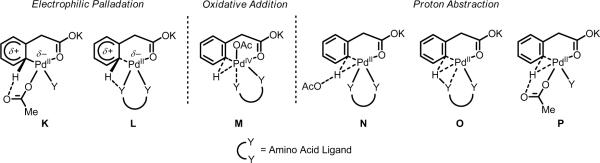
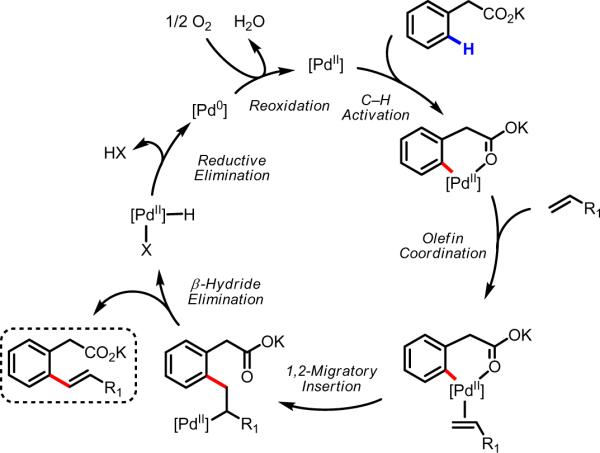
Initial rate studies have revealed dramatic acceleration in aerobic Pd(II)-catalyzed C-H olefination reactions of phenylacetic acids when mono-N-protected amino acids are used as ligands. In light of these findings, systematic ligand tuning was undertaken, which has resulted in drastic improvements in substrate scope, reaction rate, and catalyst turnover. We present evidence from intermolecular competition studies and kinetic isotope effect experiments that implies that the observed rate increases are a result of acceleration in the C-H cleavage step. Furthermore, these studies suggest that the origin of this phenomenon is a change in the mechanism of C-H cleavage from electrophilic palladation to proton abstraction.
Figures
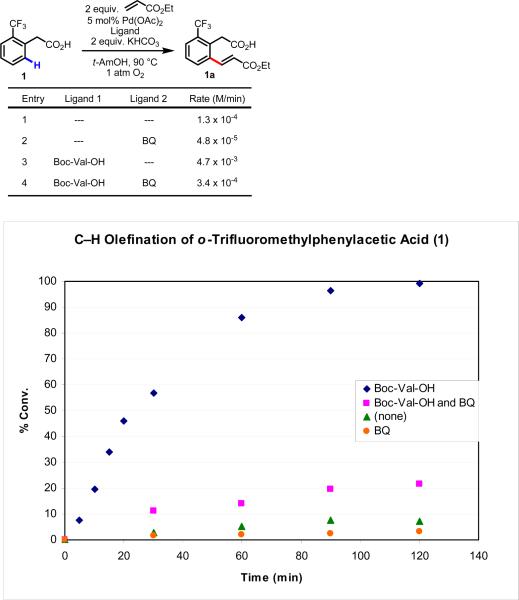
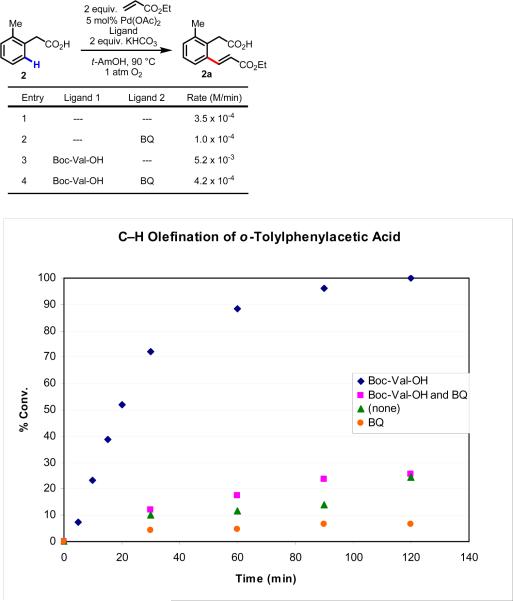
References
-
-
For the initial reports of the Mizoroki–Heck reaction, see: Mizoroki T, Mori K, Ozaki A. Bull. Chem. Soc. Jpn. 1971;44:581.. Heck RF, Nolley JP., Jr. J. Org. Chem. 1972;37:2320..
-
For reviews, see: Heck RF. Acc. Chem. Res. 1979;12:146.. Heck RF. Org. React. 1982;27:345.. Tsuji J. Palladium Reagents and Catalysts. Wiley; Chichester, UK: 1995. Chapter 4.. Bräse S, de Meijere A. Chapter 5. In: de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; New York: 2004. . Dounay AB, Overman LE. Chem. Rev. 2003;103:2945..
-
-
-
For the first reports of Pd(0)-catalyzed aryl halide/alkyne coupling, see: Cassar L. J. Organomet. Chem. 1975;93:253.. Dieck AH, Heck RF. J. Organomet. Chem. 1975;93:259..
-
For an improved protocol in the presence of Cu(I) salts (the Sonogashira reaction), see: Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975;16:4467..
-
-
-
For the initial reports of the Ni-catalyzed Kumada–Corriu coupling reaction, see: Corriu RJP, Masse JP. J. Chem. Soc., Chem. Commun. 1972:144a.. Tamao K, Sumitani K, Kumada M. J. Am. Chem. Soc. 1972;94:4374..
-
For the first Pd(0)-catalyzed reaction, see: Yamamura M, Moritani I, Murahashi S-I. J. Organomet. Chem. 1975;91:C39–C42..
-
-
-
For initial report of the Negishi coupling reaction, see: Negishi E, King AO, Okukado N. J. Org. Chem. 1977;42:1821..
-
For a review, see: Negishi E. Acc. Chem. Res. 1982;15:340..
-
-
-
For initial reports of the Stille coupling reaction, see: Milstein D, Stille JK. J. Am. Chem. Soc. 1978;100:3636.. Milstein D, Stille JK. J. Am. Chem. Soc. 1979;101:4992..
-
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
