

Comprehensive structural and functional characterization of the human kinome by protein structure modeling and ligand virtual screening
- PMID: 20853887
- PMCID: PMC2963673
- DOI: 10.1021/ci100235n
Comprehensive structural and functional characterization of the human kinome by protein structure modeling and ligand virtual screening
Abstract
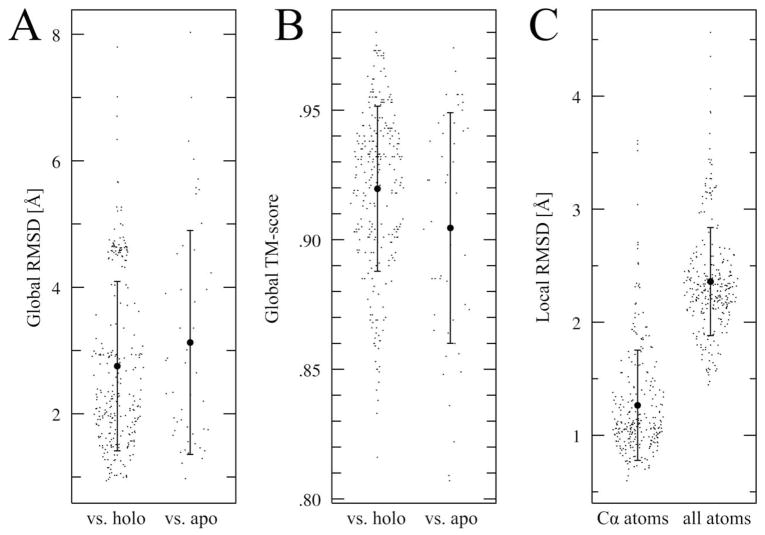
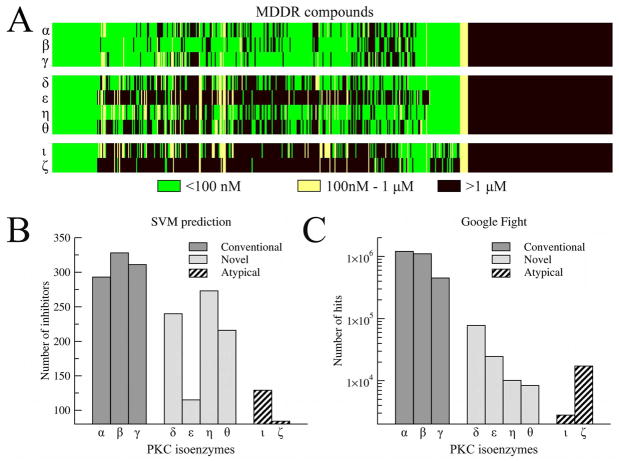
The growing interest in the identification of kinase inhibitors, promising therapeutics in the treatment of many diseases, has created a demand for the structural characterization of the entire human kinome. At the outset of the drug development process, the lead-finding stage, approaches that enrich the screening library with bioactive compounds are needed. Here, protein structure based methods can play an important role, but despite structural genomics efforts, it is unlikely that the three-dimensional structures of the entire kinome will be available soon. Therefore, at the proteome level, structure-based approaches must rely on predicted models, with a key issue being their utility in virtual ligand screening. In this study, we employ the recently developed FINDSITE/Q-Dock ligand homology modeling approach, which is well-suited for proteome-scale applications using predicted structures, to provide extensive structural and functional characterization of the human kinome. Specifically, we construct structure models for the human kinome; these are subsequently subject to virtual screening against a library of more than 2 million compounds. To rank the compounds, we employ a hierarchical approach that combines ligand- and structure-based filters. Modeling accuracy is carefully validated using available experimental data with particularly encouraging results found for the ability to identify, without prior knowledge, specific kinase inhibitors. More generally, the modeling procedure results in a large number of predicted molecular interactions between kinases and small ligands that should be of practical use in the development of novel inhibitors. The data set is freely available to the academic community via a user-friendly Web interface at http://cssb.biology.gatech.edu/kinomelhm/ as well as at the ZINC Web site ( http://zinc.docking.org/applications/2010Apr/Brylinski-2010.tar.gz ).
Figures










Similar articles
-
FINDSITE(comb): a threading/structure-based, proteomic-scale virtual ligand screening approach.J Chem Inf Model. 2013 Jan 28;53(1):230-40. doi: 10.1021/ci300510n. Epub 2012 Dec 28. J Chem Inf Model. 2013. PMID: 23240691 Free PMC article.
-
Cross-reactivity virtual profiling of the human kinome by X-react(KIN): a chemical systems biology approach.Mol Pharm. 2010 Dec 6;7(6):2324-33. doi: 10.1021/mp1002976. Epub 2010 Nov 8. Mol Pharm. 2010. PMID: 20958088 Free PMC article.
-
Nonlinear scoring functions for similarity-based ligand docking and binding affinity prediction.J Chem Inf Model. 2013 Nov 25;53(11):3097-112. doi: 10.1021/ci400510e. Epub 2013 Nov 11. J Chem Inf Model. 2013. PMID: 24171431
-
Protein kinase homology models: recent developments and results.Curr Med Chem. 2011;18(19):2848-53. doi: 10.2174/092986711796150441. Curr Med Chem. 2011. PMID: 21651494 Review.
-
Virtual screening for kinase targets.Curr Med Chem. 2004 Mar;11(6):693-707. doi: 10.2174/0929867043455684. Curr Med Chem. 2004. PMID: 15032724 Review.
Cited by
-
Structure-based virtual screening and biological evaluation of novel small-molecule BTK inhibitors.J Enzyme Inhib Med Chem. 2022 Dec;37(1):226-235. doi: 10.1080/14756366.2021.1999237. J Enzyme Inhib Med Chem. 2022. PMID: 34894949 Free PMC article.
-
FINDSITEcomb2.0: A New Approach for Virtual Ligand Screening of Proteins and Virtual Target Screening of Biomolecules.J Chem Inf Model. 2018 Nov 26;58(11):2343-2354. doi: 10.1021/acs.jcim.8b00309. Epub 2018 Oct 16. J Chem Inf Model. 2018. PMID: 30278128 Free PMC article.
-
Are predicted protein structures of any value for binding site prediction and virtual ligand screening?Curr Opin Struct Biol. 2013 Apr;23(2):191-7. doi: 10.1016/j.sbi.2013.01.009. Epub 2013 Feb 14. Curr Opin Struct Biol. 2013. PMID: 23415854 Free PMC article. Review.
-
Kinase Atlas: Druggability Analysis of Potential Allosteric Sites in Kinases.J Med Chem. 2019 Jul 25;62(14):6512-6524. doi: 10.1021/acs.jmedchem.9b00089. Epub 2019 Jul 5. J Med Chem. 2019. PMID: 31274316 Free PMC article. Review.
-
Predicting Ligand Binding Sites on Protein Surfaces by 3-Dimensional Probability Density Distributions of Interacting Atoms.PLoS One. 2016 Aug 11;11(8):e0160315. doi: 10.1371/journal.pone.0160315. eCollection 2016. PLoS One. 2016. PMID: 27513851 Free PMC article.
References
-
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–34. - PubMed
-
- Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9(8):576–96. - PubMed
-
- Kennelly PJ. Protein kinases and protein phosphatases in prokaryotes: a genomic perspective. FEMS Microbiol Lett. 2002;206(1):1–8. - PubMed
-
- Adcock IM, Chung KF, Caramori G, Ito K. Kinase inhibitors and airway inflammation. Eur J Pharmacol. 2006;533(1–3):118–32. - PubMed
-
- Basu A. The potential of protein kinase C as a target for anticancer treatment. Pharmacol Ther. 1993;59(3):257–80. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
