

The tarantula toxins ProTx-II and huwentoxin-IV differentially interact with human Nav1.7 voltage sensors to inhibit channel activation and inactivation
- PMID: 20855463
- PMCID: PMC2993464
- DOI: 10.1124/mol.110.066332
The tarantula toxins ProTx-II and huwentoxin-IV differentially interact with human Nav1.7 voltage sensors to inhibit channel activation and inactivation
Abstract
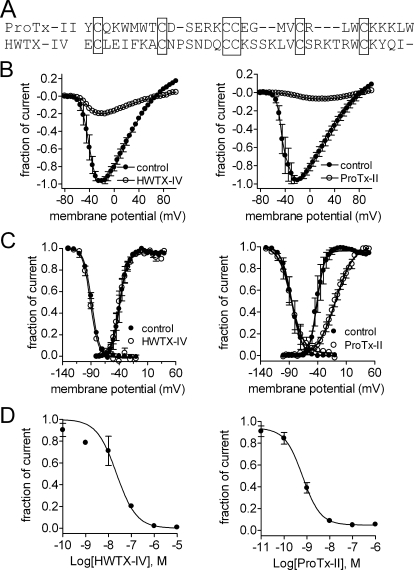
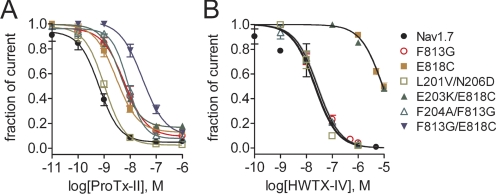
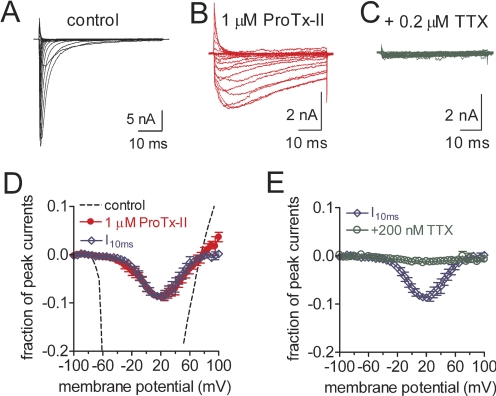
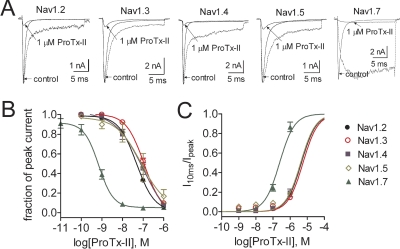
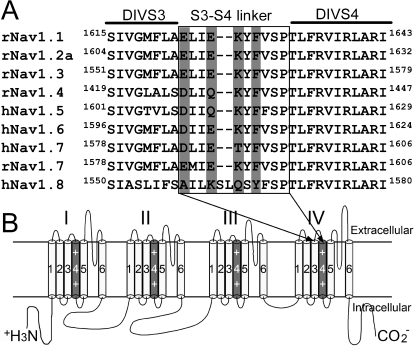
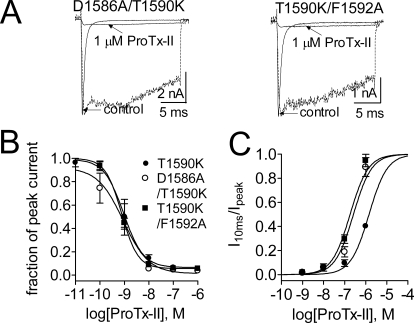
The voltage-gated sodium channel Na(v)1.7 plays a crucial role in pain, and drugs that inhibit hNa(v)1.7 may have tremendous therapeutic potential. ProTx-II and huwentoxin-IV (HWTX-IV), cystine knot peptides from tarantula venoms, preferentially block hNa(v)1.7. Understanding the interactions of these toxins with sodium channels could aid the development of novel pain therapeutics. Whereas both ProTx-II and HWTX-IV have been proposed to preferentially block hNa(v)1.7 activation by trapping the domain II voltage-sensor in the resting configuration, we show that specific residues in the voltage-sensor paddle of domain II play substantially different roles in determining the affinities of these toxins to hNa(v)1.7. The mutation E818C increases ProTx-II's and HWTX-IV's IC(50) for block of hNa(v)1.7 currents by 4- and 400-fold, respectively. In contrast, the mutation F813G decreases ProTx-II affinity by 9-fold but has no effect on HWTX-IV affinity. It is noteworthy that we also show that ProTx-II, but not HWTX-IV, preferentially interacts with hNa(v)1.7 to impede fast inactivation by trapping the domain IV voltage-sensor in the resting configuration. Mutations E1589Q and T1590K in domain IV each decreased ProTx-II's IC(50) for impairment of fast inactivation by ~6-fold. In contrast mutations D1586A and F1592A in domain-IV increased ProTx-II's IC(50) for impairment of fast inactivation by ~4-fold. Our results show that whereas ProTx-II and HWTX-IV binding determinants on domain-II may overlap, domain II plays a much more crucial role for HWTX-IV, and contrary to what has been proposed to be a guiding principle of sodium channel pharmacology, molecules do not have to exclusively target the domain IV voltage-sensor to influence sodium channel inactivation.
Figures

References
-
- Catterall WA, Goldin AL, Waxman SGInternational Union of Pharmacology (2003) International Union of Pharmacology. XXXIX. Compendium of voltage-gated ion channels: sodium channels. Pharmacol Rev 55:575–578 - PubMed
-
- Cestèle S, Catterall WA. (2000) Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 82:883–892 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
