

Whole body deletion of AMP-activated protein kinase {beta}2 reduces muscle AMPK activity and exercise capacity
- PMID: 20855892
- PMCID: PMC2988326
- DOI: 10.1074/jbc.M110.102434
Whole body deletion of AMP-activated protein kinase {beta}2 reduces muscle AMPK activity and exercise capacity
Abstract
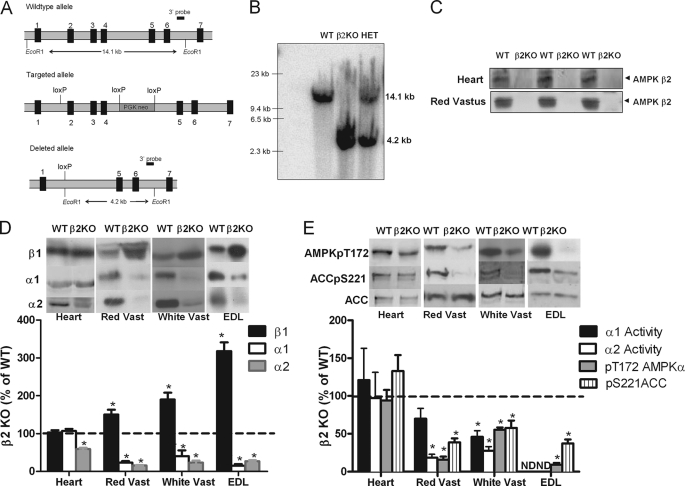
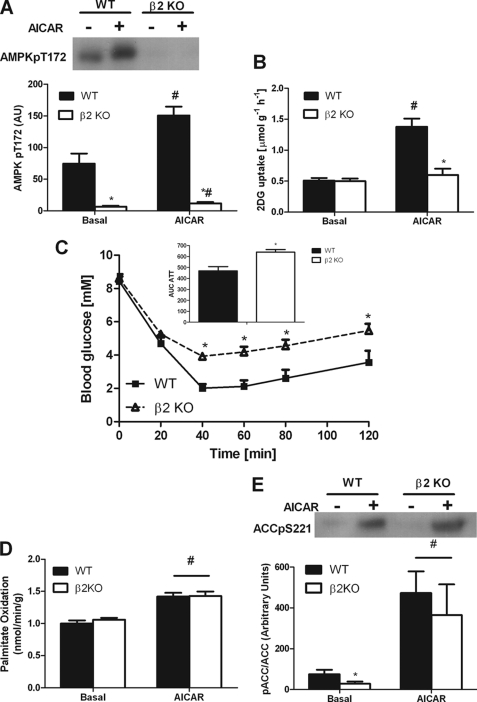
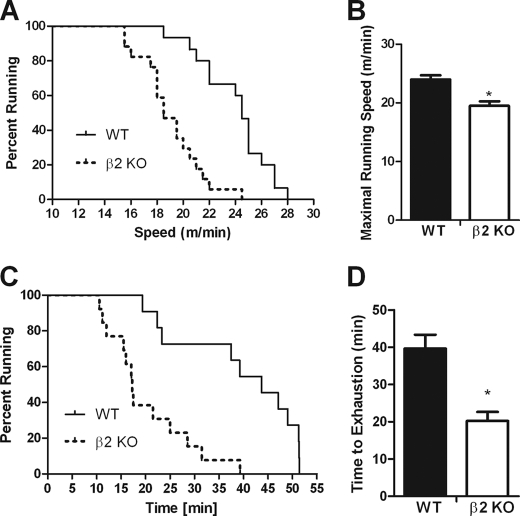
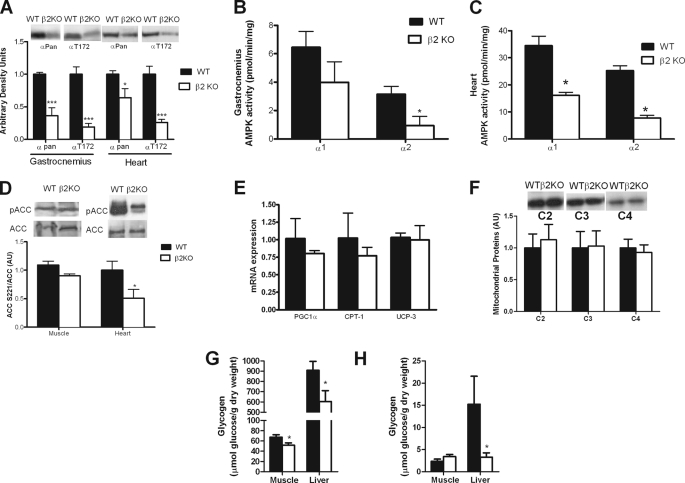
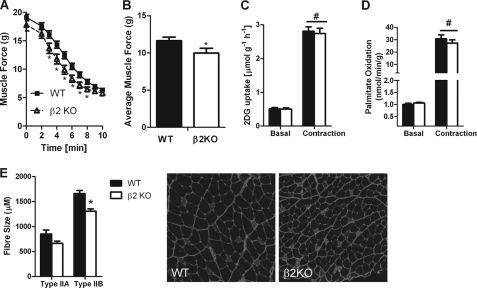
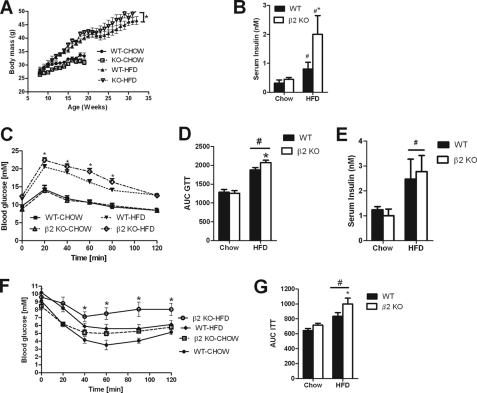
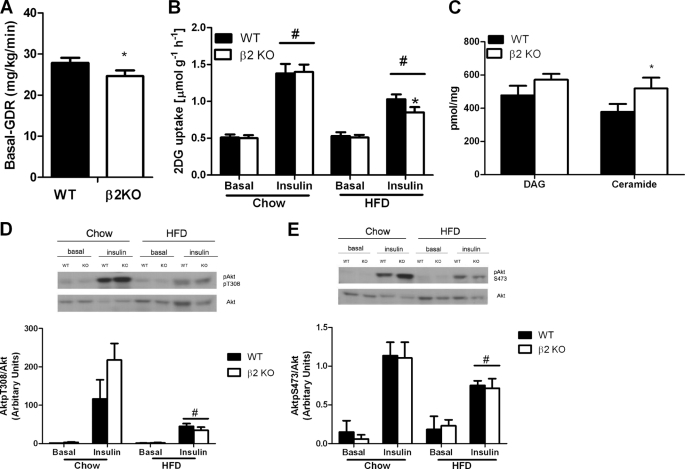
AMP-activated protein kinase (AMPK) β subunits (β1 and β2) provide scaffolds for binding α and γ subunits and contain a carbohydrate-binding module important for regulating enzyme activity. We generated C57Bl/6 mice with germline deletion of AMPK β2 (β2 KO) and examined AMPK expression and activity, exercise capacity, metabolic control during muscle contractions, aminoimidazole carboxamide ribonucleotide (AICAR) sensitivity, and susceptibility to obesity-induced insulin resistance. We find that β2 KO mice are viable and breed normally. β2 KO mice had a reduction in skeletal muscle AMPK α1 and α2 expression despite up-regulation of the β1 isoform. Heart AMPK α2 expression was also reduced but this did not affect resting AMPK α1 or α2 activities. AMPK α1 and α2 activities were not changed in liver, fat, or hypothalamus. AICAR-stimulated glucose uptake but not fatty acid oxidation was impaired in β2 KO mice. During treadmill running β2 KO mice had reduced maximal and endurance exercise capacity, which was associated with lower muscle and heart AMPK activity and reduced levels of muscle and liver glycogen. Reductions in exercise capacity of β2 KO mice were not due to lower muscle mitochondrial content or defects in contraction-stimulated glucose uptake or fatty acid oxidation. When challenged with a high-fat diet β2 KO mice gained more weight and were more susceptible to the development of hyperinsulinemia and glucose intolerance. In summary these data show that deletion of AMPK β2 reduces AMPK activity in skeletal muscle resulting in impaired exercise capacity and the worsening of diet-induced obesity and glucose intolerance.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
