

Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment
- PMID: 20855962
- PMCID: PMC3047617
- DOI: 10.4161/cc.9.17.12928
Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment
Abstract
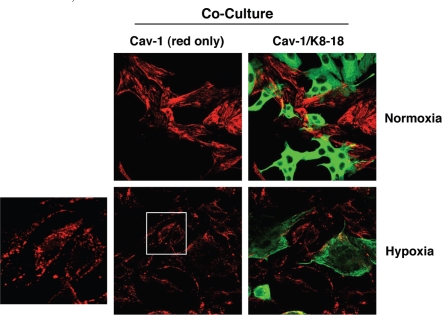
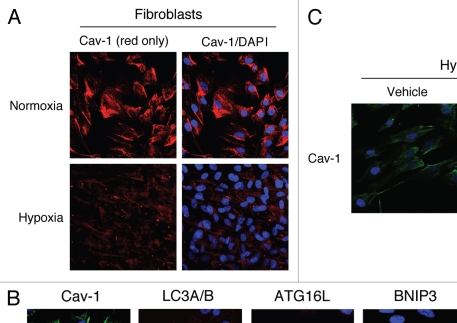
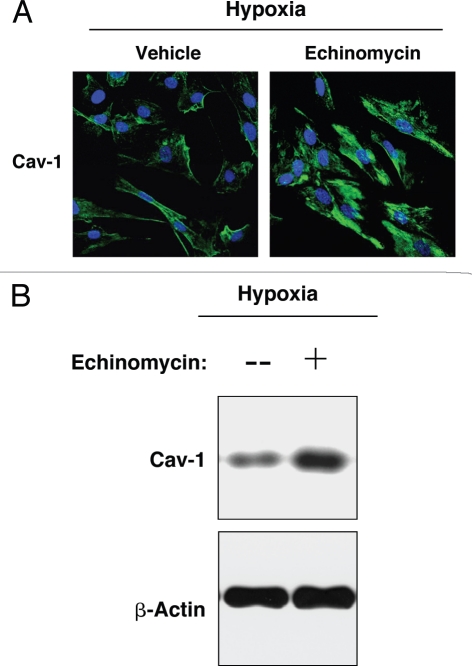
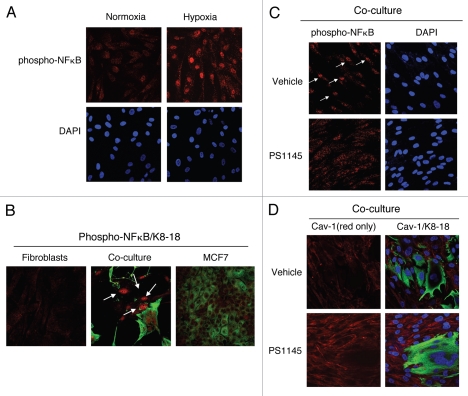
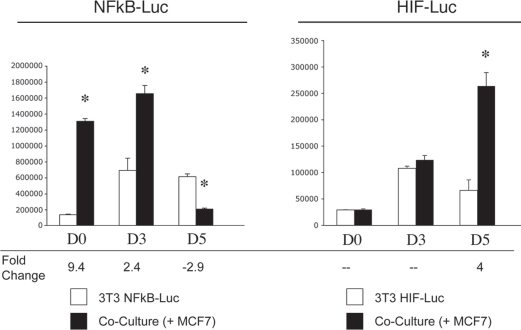
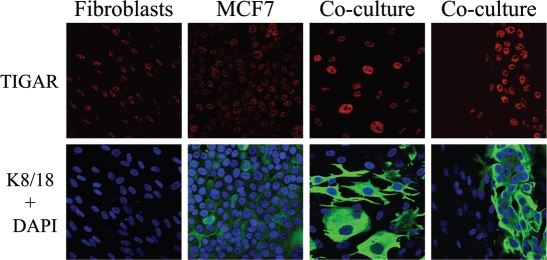
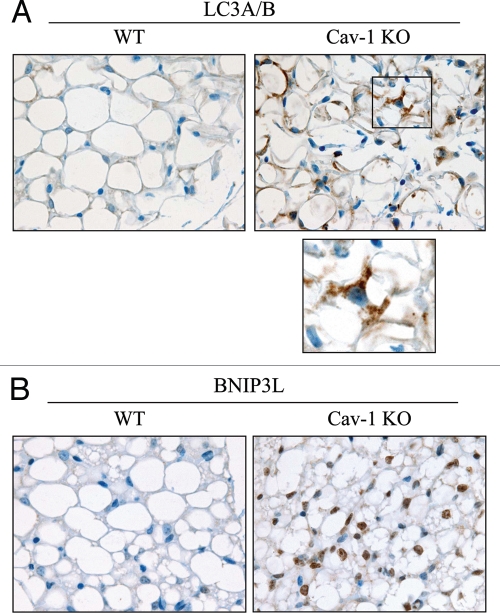
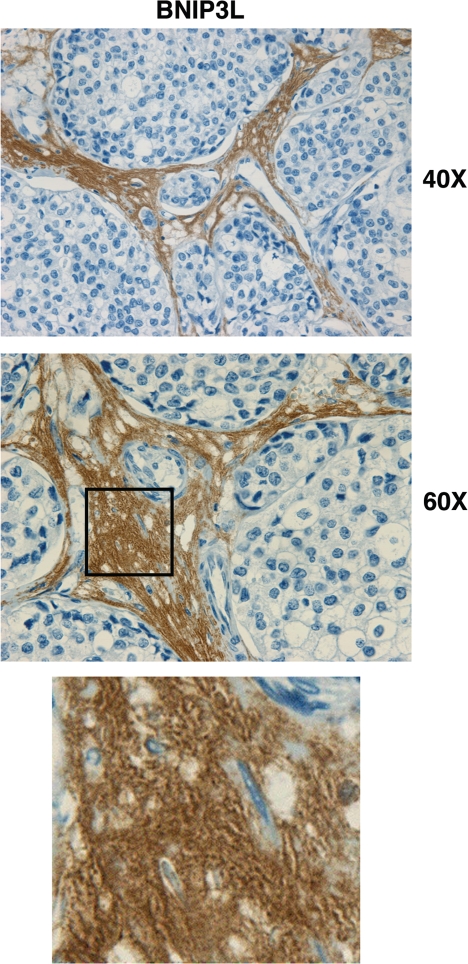
Recently, using a co-culture system, we demonstrated that MCF7 epithelial cancer cells induce oxidative stress in adjacent cancer-associated fibroblasts, resulting in the autophagic/lysosomal degradation of stromal caveolin-1 (Cav-1). However, the detailed signaling mechanism(s) underlying this process remain largely unknown. Here, we show that hypoxia is sufficient to induce the autophagic degradation of Cav-1 in stromal fibroblasts, which is blocked by the lysosomal inhibitor chloroquine. Concomitant with the hypoxia-induced degradation of Cav-1, we see the upregulation of a number of well-established autophagy/mitophagy markers, namely LC3, ATG16L, BNIP3, BNIP3L, HIF-1α and NFκB. In addition, pharmacological activation of HIF-1α drives Cav-1 degradation, while pharmacological inactivation of HIF-1 prevents the downregulation of Cav-1. Similarly, pharmacological inactivation of NFκB--another inducer of autophagy-prevents Cav-1 degradation. Moreover, treatment with an inhibitor of glutathione synthase, namely BSO, which induces oxidative stress via depletion of the reduced glutathione pool, is sufficient to induce the autophagic degradation of Cav-1. Thus, it appears that oxidative stress mediated induction of HIF1- and NFκB-activation in fibroblasts drives the autophagic degradation of Cav-1. In direct support of this hypothesis, we show that MCF7 cancer cells activate HIF-1α- and NFκB-driven luciferase reporters in adjacent cancer-associated fibroblasts, via a paracrine mechanism. Consistent with these findings, acute knock-down of Cav-1 in stromal fibroblasts, using an siRNA approach, is indeed sufficient to induce autophagy, with the upregulation of both lysosomal and mitophagy markers. How does the loss of stromal Cav-1 and the induction of stromal autophagy affect cancer cell survival? Interestingly, we show that a loss of Cav-1 in stromal fibroblasts protects adjacent cancer cells against apoptotic cell death. Thus, autophagic cancer-associated fibroblasts, in addition to providing recycled nutrients for cancer cell metabolism, also play a protective role in preventing the death of adjacent epithelial cancer cells. We demonstrate that cancer-associated fibroblasts upregulate the expression of TIGAR in adjacent epithelial cancer cells, thereby conferring resistance to apoptosis and autophagy. Finally, the mammary fat pads derived from Cav-1 (-/-) null mice show a hypoxia-like response in vivo, with the upregulation of autophagy markers, such as LC3 and BNIP3L. Taken together, our results provide direct support for the "Autophagic Tumor Stroma Model of Cancer Metabolism", and explain the exceptional prognostic value of a loss of stromal Cav-1 in cancer patients. Thus, a loss of stromal fibroblast Cav-1 is a biomarker for chronic hypoxia, oxidative stress and autophagy in the tumor microenvironment, consistent with its ability to predict early tumor recurrence, lymph node metastasis and tamoxifen-resistance in human breast cancers. Our results imply that cancer patients lacking stromal Cav-1 should benefit from HIF-inhibitors, NFκB-inhibitors, anti-oxidant therapies, as well as autophagy/lysosomal inhibitors. These complementary targeted therapies could be administered either individually or in combination, to prevent the onset of autophagy in the tumor stromal compartment, which results in a "lethal" tumor microenvironment.
Figures








References
-
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. - PubMed
-
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. - PubMed
-
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997;272:22642–22647. - PubMed
-
- Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. - PubMed
-
- Schofield CJ, Ratcliffe PJ. Signalling hypoxia by HIF hydroxylases. Biochem Biophys Res Commun. 2005;338:617–626. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA075503/CA/NCI NIH HHS/United States
- R01 CA098779/CA/NCI NIH HHS/United States
- R01-CA-120876/CA/NCI NIH HHS/United States
- R01 CA120876/CA/NCI NIH HHS/United States
- R01-CA-70896/CA/NCI NIH HHS/United States
- R01-CA-098779/CA/NCI NIH HHS/United States
- R01-CA-86072/CA/NCI NIH HHS/United States
- R01-AR-055660/AR/NIAMS NIH HHS/United States
- R01-CA-080250/CA/NCI NIH HHS/United States
- R01 CA070896/CA/NCI NIH HHS/United States
- R01 CA107382/CA/NCI NIH HHS/United States
- P30 CA056036/CA/NCI NIH HHS/United States
- P30-CA-56036/CA/NCI NIH HHS/United States
- R01-CA-107382/CA/NCI NIH HHS/United States
- R01 AR055660/AR/NIAMS NIH HHS/United States
- R01-CA-75503/CA/NCI NIH HHS/United States
- R01 CA080250/CA/NCI NIH HHS/United States
- R01 CA086072/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical