

Structural Survey of Zinc Containing Proteins and the Development of the Zinc AMBER Force Field (ZAFF)
- PMID: 20856692
- PMCID: PMC2941202
- DOI: 10.1021/ct1002626
Structural Survey of Zinc Containing Proteins and the Development of the Zinc AMBER Force Field (ZAFF)
Abstract
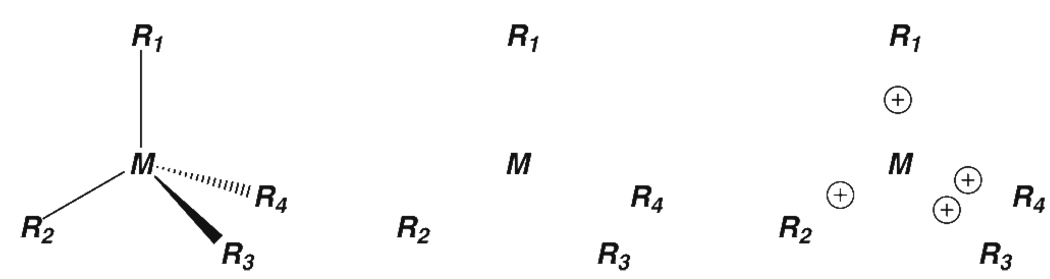
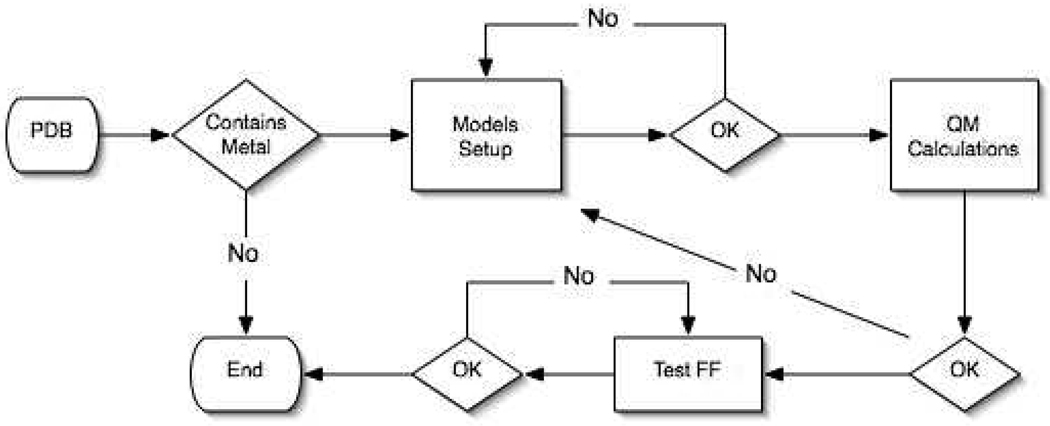
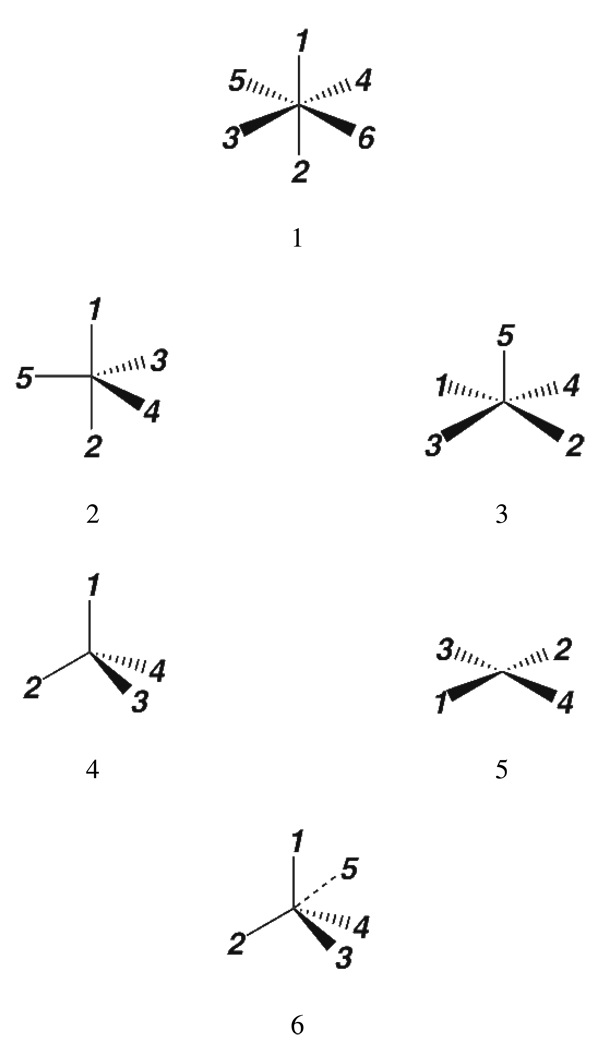
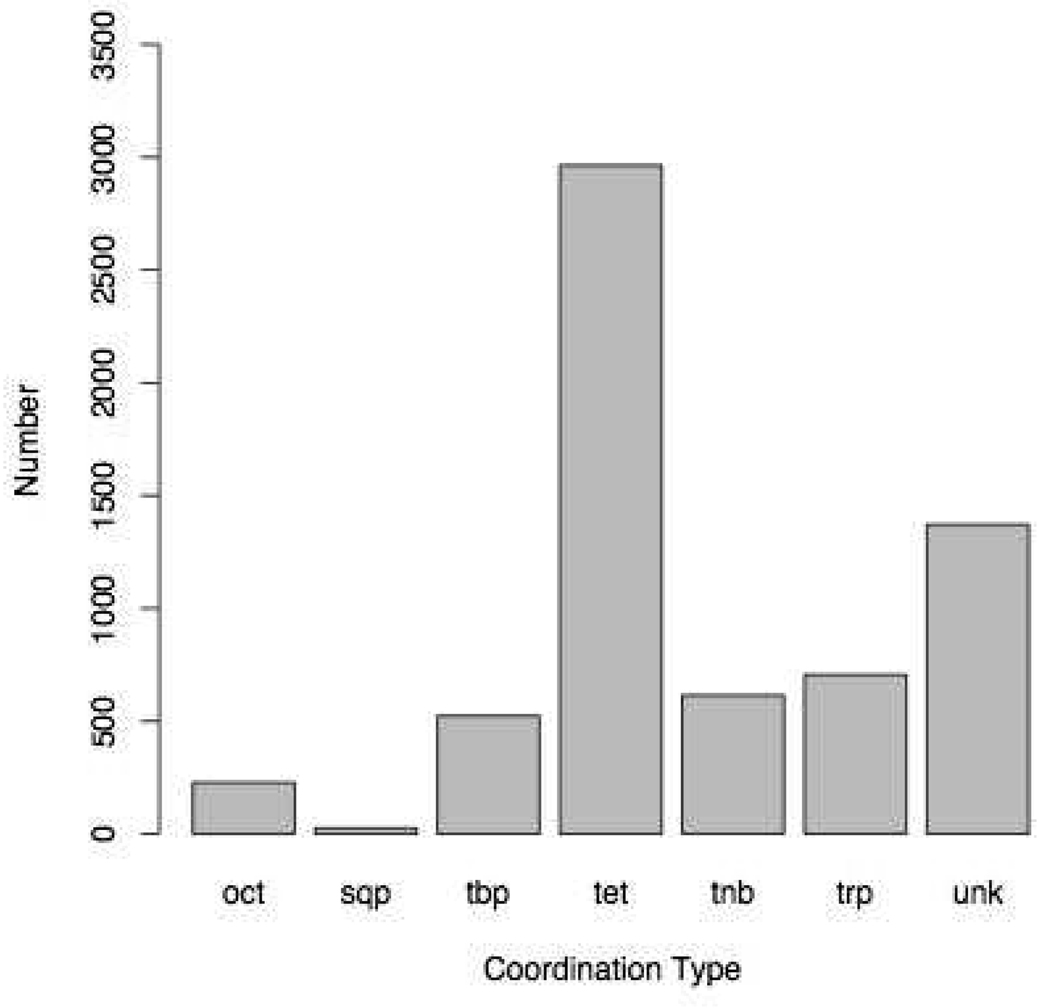
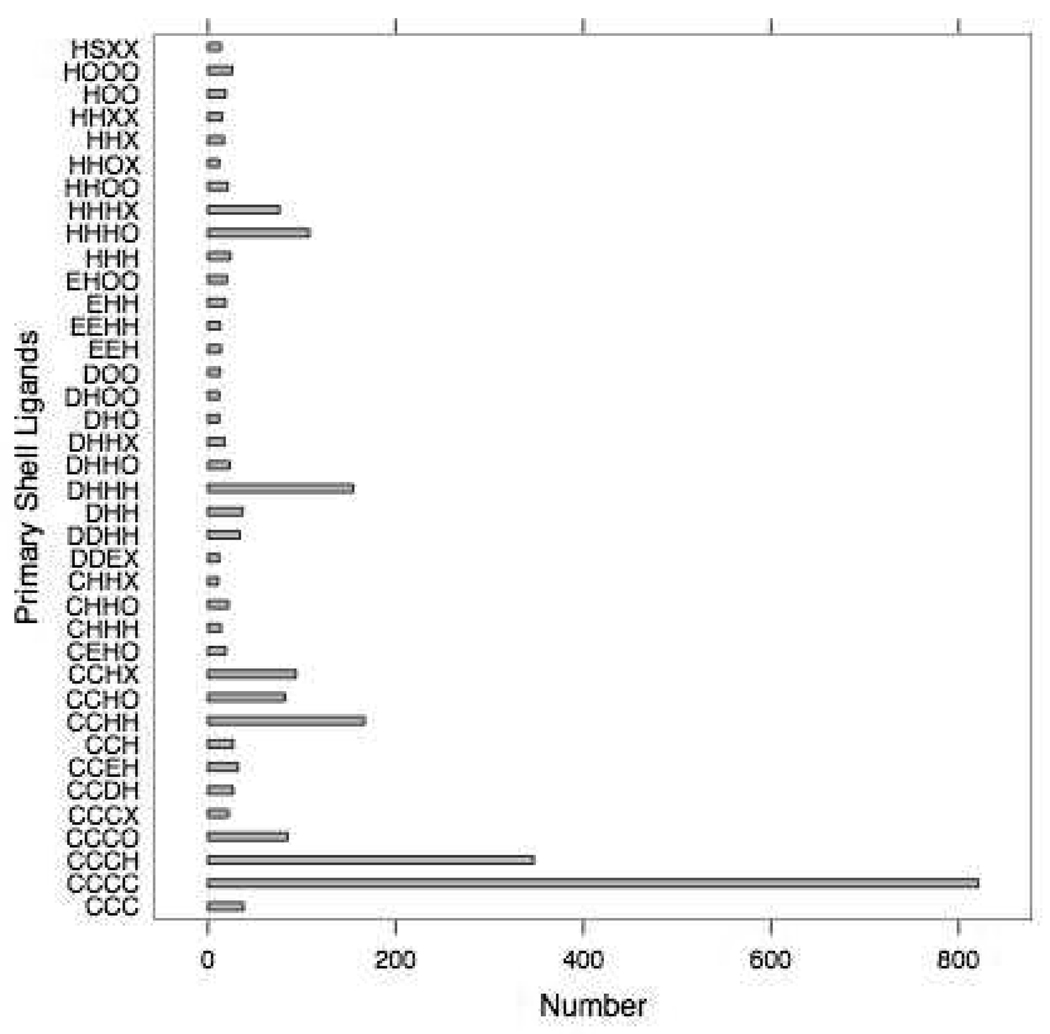
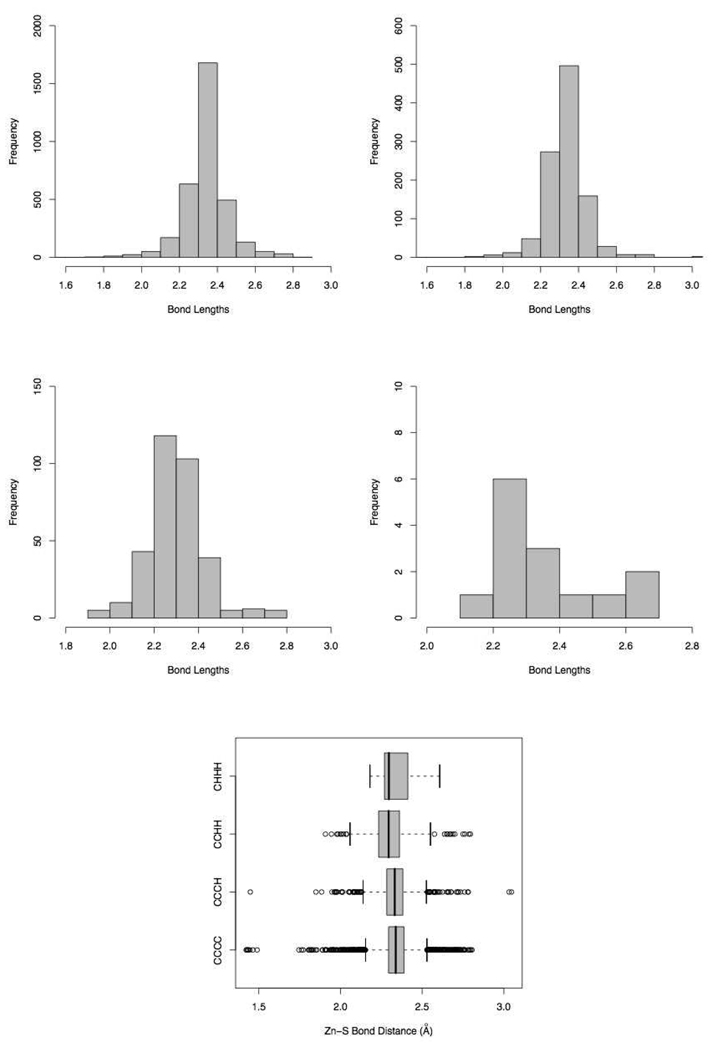
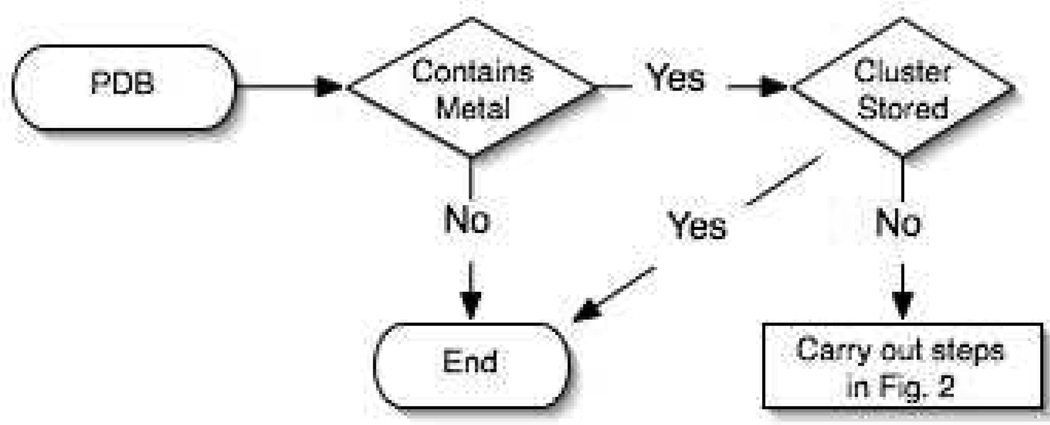
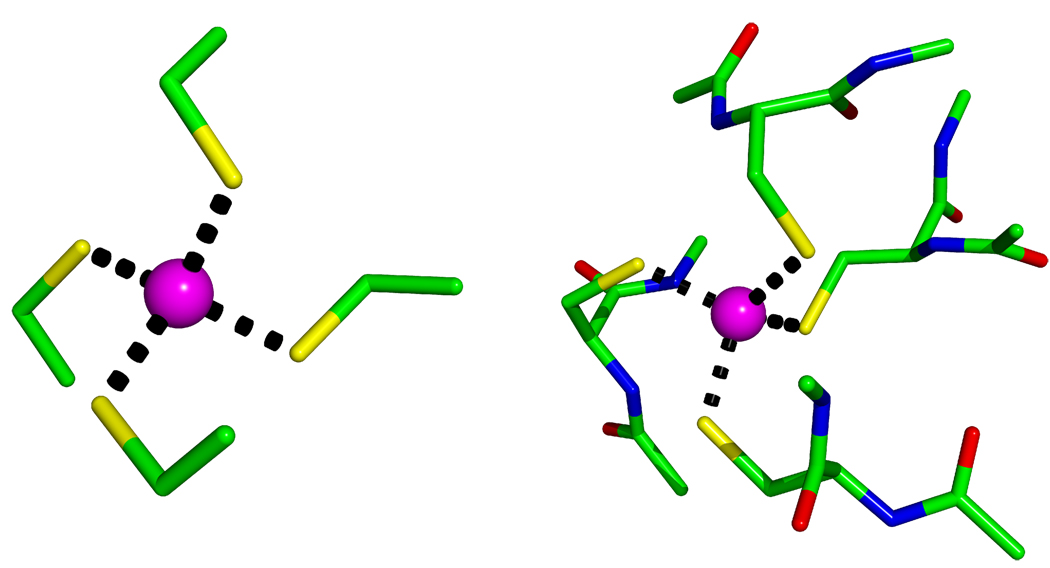
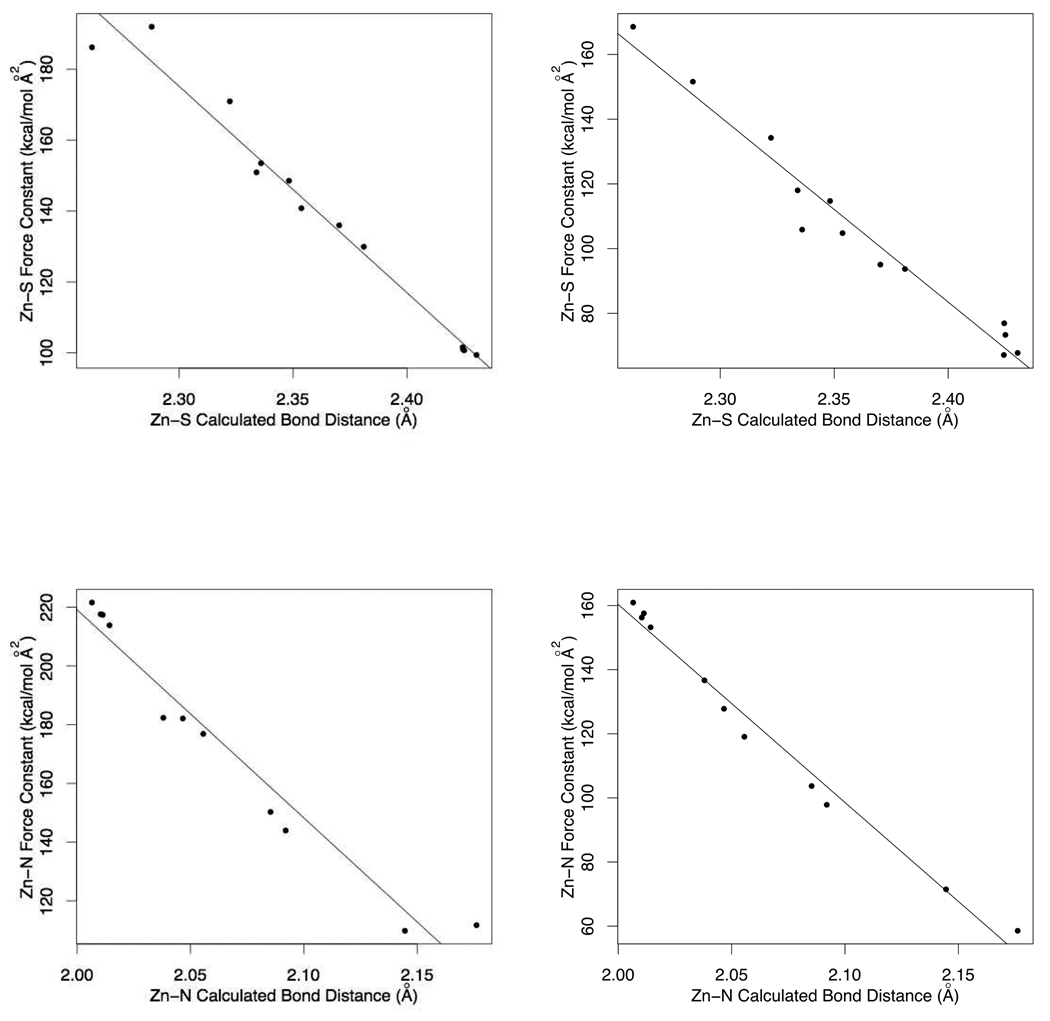
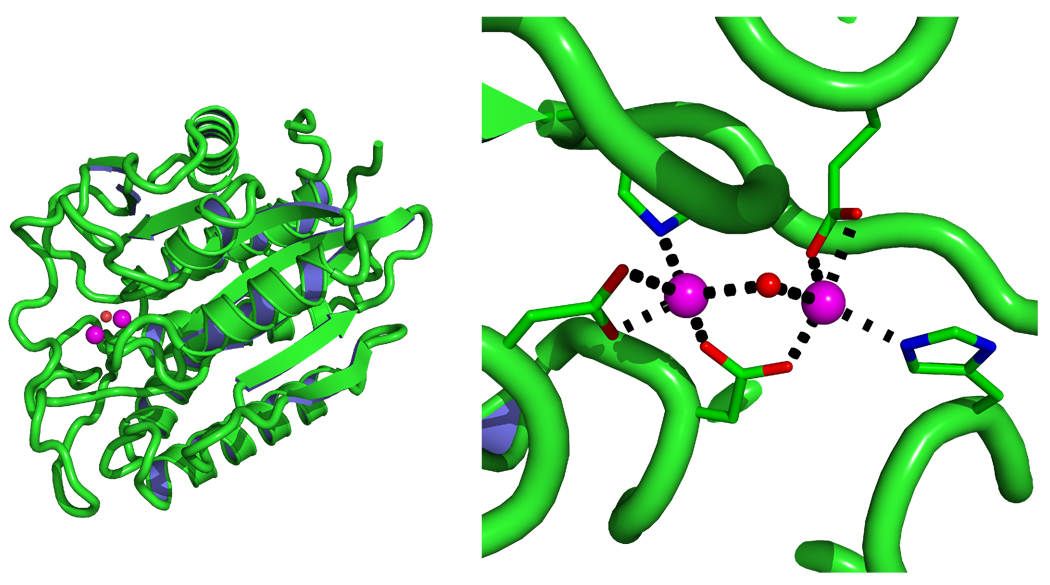
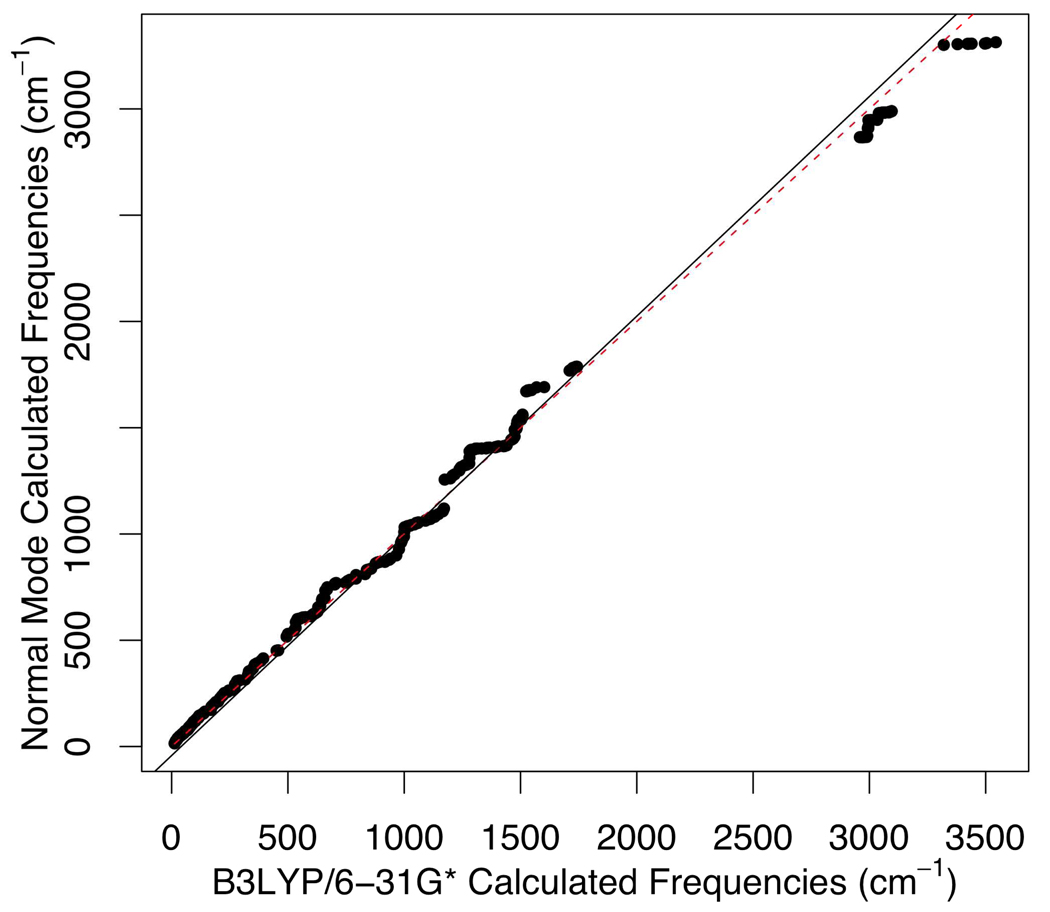
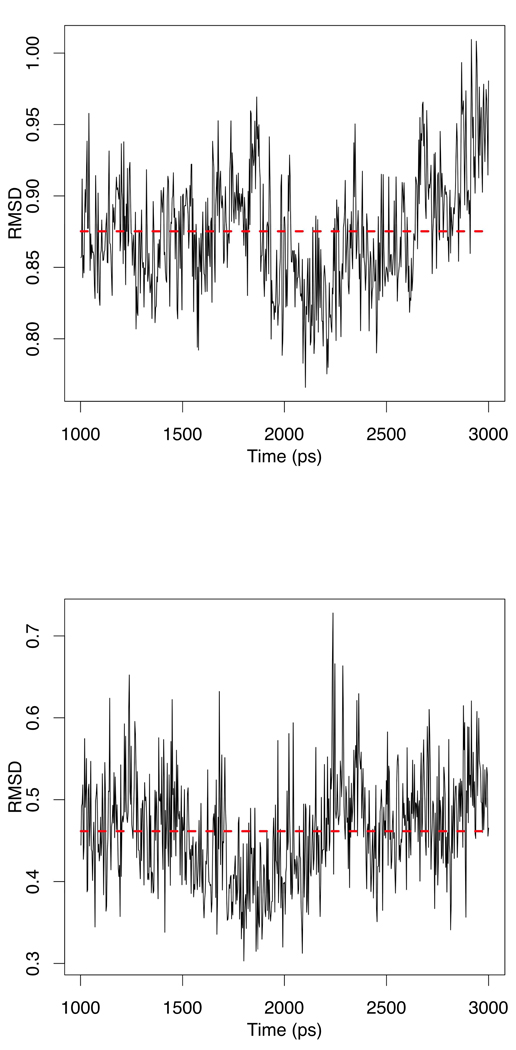
Currently the Protein Data Bank (PDB) contains over 18,000 structures that contain a metal ion including Na, Mg, K, Ca, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Pd, Ag, Cd, Ir, Pt, Au, and Hg. In general, carrying out classical molecular dynamics (MD) simulations of metalloproteins is a convoluted and time consuming process. Herein, we describe MCPB (Metal Center Parameter Builder), which allows one, to conveniently and rapidly incorporate metal ions using the bonded plus electrostatics model (Hoops et al., J. Am. Chem. Soc. 1991, 113, 8262-8270) into the AMBER Force Field (FF). MCPB was used to develop a Zinc FF, ZAFF, which is compatible with the existing AMBER FFs. The PDB was mined for all Zn containing structures with most being tetrahedrally bound. The most abundant primary shell ligand combinations were extracted and FFs were created. These include Zn bound to CCCC, CCCH, CCHH, CHHH, HHHH, HHHO, HHOO, HOOO, HHHD, and HHDD (O = water and the remaining are 1 letter amino acid codes). Bond and angle force constants and RESP charges were obtained from B3LYP/6-31G* calculations of model structures from the various primary shell combinations. MCPB and ZAFF can be used to create FFs for MD simulations of metalloproteins to study enzyme catalysis, drug design and metalloprotein crystal refinement.
Figures
Similar articles
-
A Comparison of Bonded and Nonbonded Zinc(II) Force Fields with NMR Data.Int J Mol Sci. 2023 Mar 13;24(6):5440. doi: 10.3390/ijms24065440. Int J Mol Sci. 2023. PMID: 36982515 Free PMC article.
-
MCPB.py: A Python Based Metal Center Parameter Builder.J Chem Inf Model. 2016 Apr 25;56(4):599-604. doi: 10.1021/acs.jcim.5b00674. Epub 2016 Mar 23. J Chem Inf Model. 2016. PMID: 26913476
-
A structure-based analysis of the vibrational spectra of nitrosyl ligands in transition-metal coordination complexes and clusters.Spectrochim Acta A Mol Biomol Spectrosc. 2011 Jan;78(1):7-28. doi: 10.1016/j.saa.2010.08.001. Epub 2010 Aug 17. Spectrochim Acta A Mol Biomol Spectrosc. 2011. PMID: 21123107
-
Biomolecular force fields: where have we been, where are we now, where do we need to go and how do we get there?J Comput Aided Mol Des. 2019 Feb;33(2):133-203. doi: 10.1007/s10822-018-0111-4. Epub 2018 Nov 30. J Comput Aided Mol Des. 2019. PMID: 30506158 Review.
-
Force field development phase II: Relaxation of physics-based criteria… or inclusion of more rigorous physics into the representation of molecular energetics.J Comput Aided Mol Des. 2019 Feb;33(2):205-264. doi: 10.1007/s10822-018-0134-x. Epub 2018 Nov 30. J Comput Aided Mol Des. 2019. PMID: 30506159 Review.
Cited by
-
Nucleotide-dependent mechanism of Get3 as elucidated from free energy calculations.Proc Natl Acad Sci U S A. 2012 May 15;109(20):7759-64. doi: 10.1073/pnas.1117441109. Epub 2012 Apr 30. Proc Natl Acad Sci U S A. 2012. PMID: 22547793 Free PMC article.
-
Computed Protein-Protein Enthalpy Signatures as a Tool for Identifying Conformation Sampling Problems.J Chem Inf Model. 2023 Oct 9;63(19):6095-6108. doi: 10.1021/acs.jcim.3c01041. Epub 2023 Sep 27. J Chem Inf Model. 2023. PMID: 37759363 Free PMC article.
-
Development of CHARMM-Compatible Force-Field Parameters for Cobalamin and Related Cofactors from Quantum Mechanical Calculations.J Chem Theory Comput. 2018 Feb 13;14(2):784-798. doi: 10.1021/acs.jctc.7b01236. Epub 2018 Feb 1. J Chem Theory Comput. 2018. PMID: 29334459 Free PMC article.
-
AutoDock4(Zn): an improved AutoDock force field for small-molecule docking to zinc metalloproteins.J Chem Inf Model. 2014 Aug 25;54(8):2371-9. doi: 10.1021/ci500209e. Epub 2014 Jul 18. J Chem Inf Model. 2014. PMID: 24931227 Free PMC article.
-
Activity prediction of substrates in NADH-dependent carbonyl reductase by docking requires catalytic constraints and charge parameterization of catalytic zinc environment.J Comput Aided Mol Des. 2015 Nov;29(11):1057-69. doi: 10.1007/s10822-015-9878-8. Epub 2015 Nov 3. J Comput Aided Mol Des. 2015. PMID: 26530855
References
-
- Hancock RD. Acc. Chem. Res. 1990;23:253.
-
- Hancock RD. Prog. Inorg. Chem. 1989;37:187.
-
- Hoops SC, Anderson KW, Merz KM., Jr J. Am. Chem. Soc. 1991;113:8262.
-
- Bayly CI, Cieplak P, Cornell WD, Kollman PA. J. Phys. Chem. 1993;97:10269.
-
- Li JB, Zhu TH, Cramer CJ, Truhlar DG. J. Phys. Chem. A. 1998;102:1820.
Grants and funding
LinkOut - more resources
Full Text Sources