

Molecular networks implicated in speech-related disorders: FOXP2 regulates the SRPX2/uPAR complex
- PMID: 20858596
- PMCID: PMC2989892
- DOI: 10.1093/hmg/ddq415
Molecular networks implicated in speech-related disorders: FOXP2 regulates the SRPX2/uPAR complex
Abstract
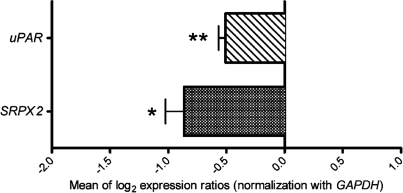
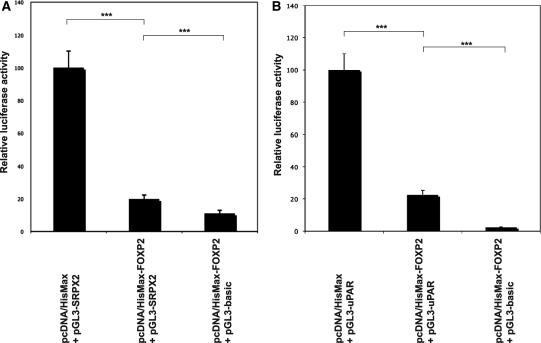
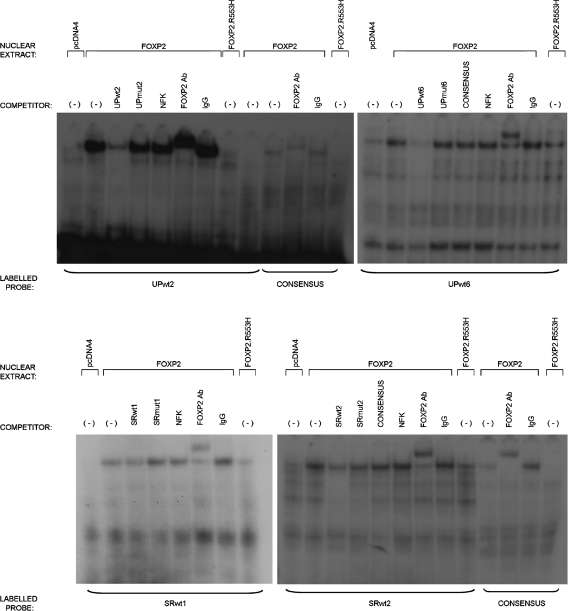
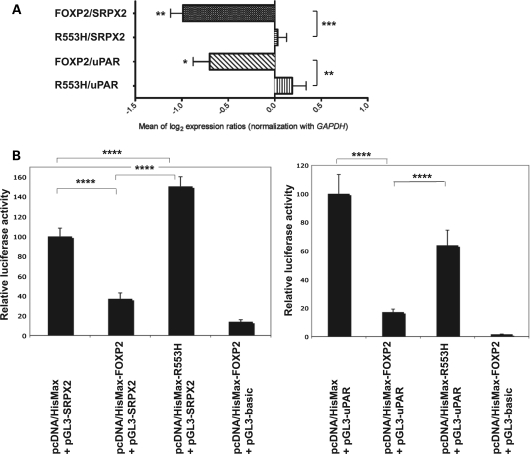
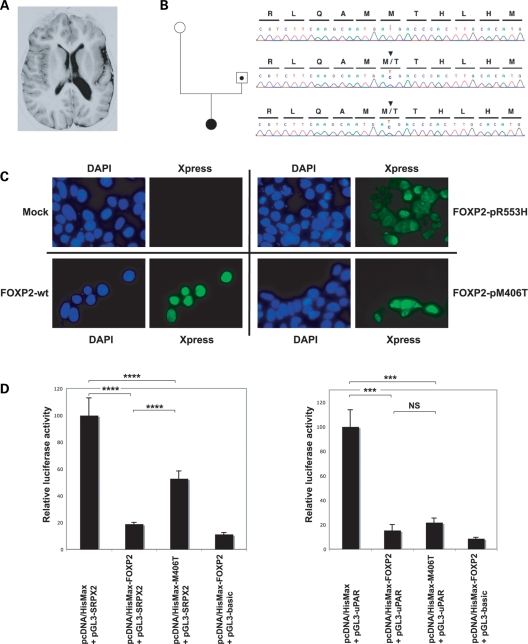
It is a challenge to identify the molecular networks contributing to the neural basis of human speech. Mutations in transcription factor FOXP2 cause difficulties mastering fluent speech (developmental verbal dyspraxia, DVD), whereas mutations of sushi-repeat protein SRPX2 lead to epilepsy of the rolandic (sylvian) speech areas, with DVD or with bilateral perisylvian polymicrogyria. Pathophysiological mechanisms driven by SRPX2 involve modified interaction with the plasminogen activator receptor (uPAR). Independent chromatin-immunoprecipitation microarray screening has identified the uPAR gene promoter as a potential target site bound by FOXP2. Here, we directly tested for the existence of a transcriptional regulatory network between human FOXP2 and the SRPX2/uPAR complex. In silico searches followed by gel retardation assays identified specific efficient FOXP2-binding sites in each of the promoter regions of SRPX2 and uPAR. In FOXP2-transfected cells, significant decreases were observed in the amounts of both SRPX2 (43.6%) and uPAR (38.6%) native transcripts. Luciferase reporter assays demonstrated that FOXP2 expression yielded a marked inhibition of SRPX2 (80.2%) and uPAR (77.5%) promoter activity. A mutant FOXP2 that causes DVD (p.R553H) failed to bind to SRPX2 and uPAR target sites and showed impaired down-regulation of SRPX2 and uPAR promoter activity. In a patient with polymicrogyria of the left rolandic operculum, a novel FOXP2 mutation (p.M406T) was found in the leucine-zipper (dimerization) domain. p.M406T partially impaired the FOXP2 regulation of SRPX2 promoter activity, whereas that of the uPAR promoter remained unchanged. Together with recently described FOXP2-CNTNAP2 and SRPX2/uPAR links, the FOXP2-SRPX2/uPAR network provides exciting insights into molecular pathways underlying speech-related disorders.
Figures
References
-
- Fisher S.E. On genes, speech, and language. N. Engl. J. Med. 2009;353:1655–1657. doi:10.1056/NEJMp058207. - DOI - PubMed
-
- Fisher S.E., Scharff C. FOXP2 as a molecular window into speech and language. Trends Genet. 2009;25:166–177. doi:10.1016/j.tig.2009.03.002. - DOI - PubMed
-
- Lai C.S., Fisher S.E., Hurst J.A., Vargha-Khadem F., Monaco A.P. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. 2001;413:519–523. doi:10.1038/35097076. - DOI - PubMed
-
- MacDermot K.D., Bonora E., Sykes N., Coupe A.M., Lai C.S., Vernes S.C., Vargha-Khadem F., McKenzie F., Smith R.L., Monaco A.P., et al. Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits. Am. J. Hum. Genet. 2005;76:1074–1080. doi:10.1086/430841. - DOI - PMC - PubMed
-
- Zeesman S., Nowaczyk M.J., Teshima I., Roberts W., Cardy J.O., Brian J., Senman L., Feuk L., Osborne L.R., Scherer S.W. Speech and language impairment and oromotor dyspraxia due to deletion of 7q31 that involves FOXP2. Am. J. Med. Genet. 2006;140:509–514. doi:10.1002/ajmg.a.31110. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
