

Accurate genome-scale percentage DNA methylation estimates from microarray data
- PMID: 20858772
- PMCID: PMC3062148
- DOI: 10.1093/biostatistics/kxq055
Accurate genome-scale percentage DNA methylation estimates from microarray data
Abstract
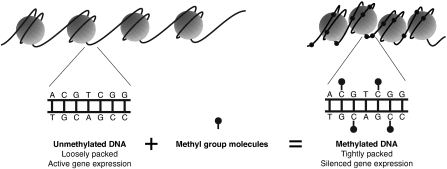
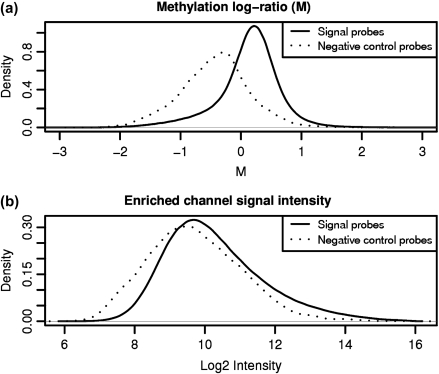
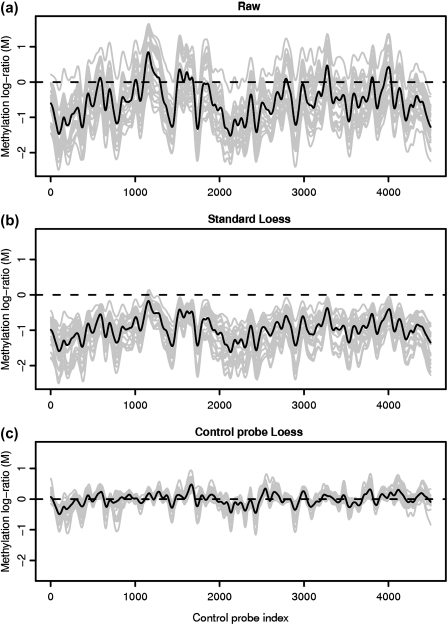
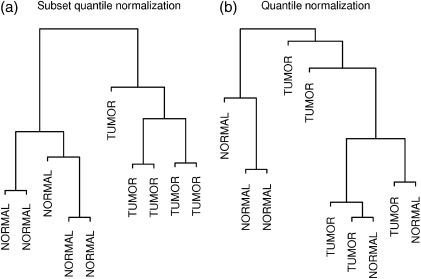
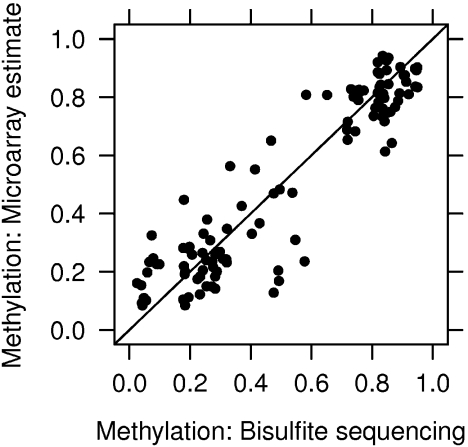
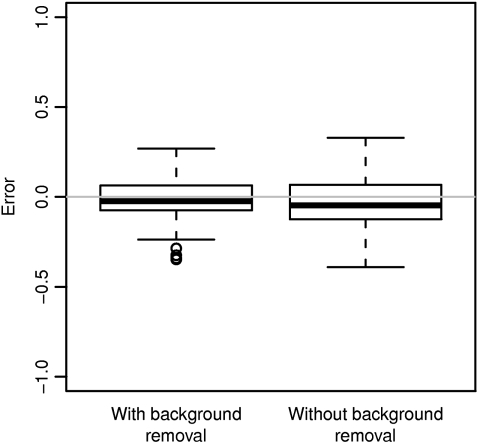
DNA methylation is a key regulator of gene function in a multitude of both normal and abnormal biological processes, but tools to elucidate its roles on a genome-wide scale are still in their infancy. Methylation sensitive restriction enzymes and microarrays provide a potential high-throughput, low-cost platform to allow methylation profiling. However, accurate absolute methylation estimates have been elusive due to systematic errors and unwanted variability. Previous microarray preprocessing procedures, mostly developed for expression arrays, fail to adequately normalize methylation-related data since they rely on key assumptions that are violated in the case of DNA methylation. We develop a normalization strategy tailored to DNA methylation data and an empirical Bayes percentage methylation estimator that together yield accurate absolute methylation estimates that can be compared across samples. We illustrate the method on data generated to detect methylation differences between tissues and between normal and tumor colon samples.
Figures
References
-
- Bird A. DNA methylation patterns and epigenetic memory. Genes and Development. 2002;16:6–21. - PubMed
-
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. - PubMed
-
- Clark SJ, Statham A, Stirzaker C, Molloy PL, Frommer M. DNA methylation: bisulphite modification and analysis. Nature Protocols. 2006;1:2353–2364. - PubMed
-
- Cloud J. Why Genes Aren't Destiny. 2010. Time. New York. Volume 175. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
