

Identification of the Drosophila ortholog of HSPB8: implication of HSPB8 loss of function in protein folding diseases
- PMID: 20858900
- PMCID: PMC2988385
- DOI: 10.1074/jbc.M110.127498
Identification of the Drosophila ortholog of HSPB8: implication of HSPB8 loss of function in protein folding diseases
Abstract
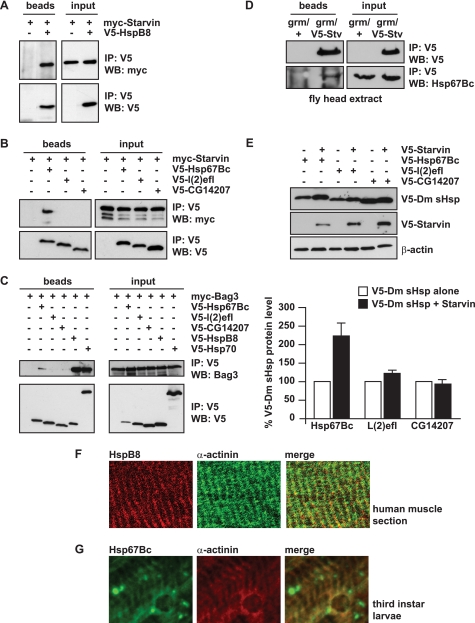
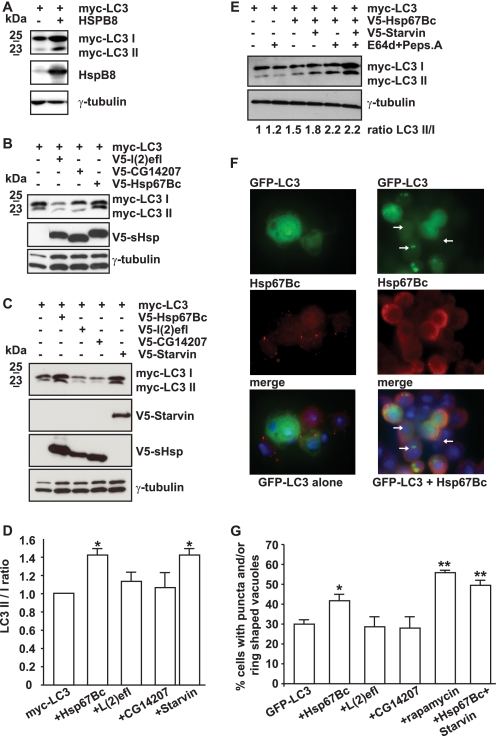
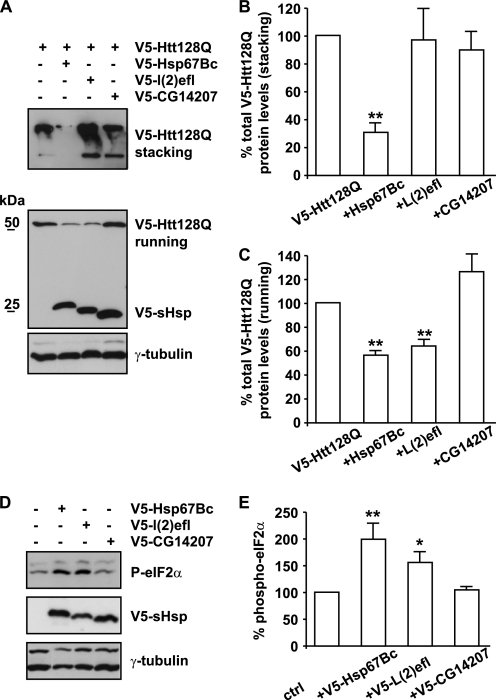
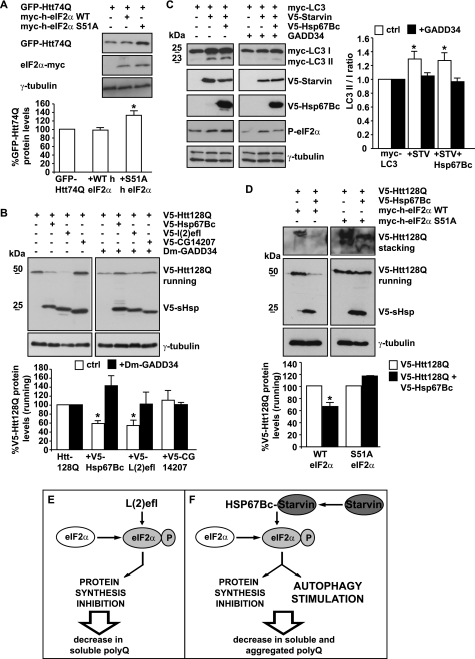
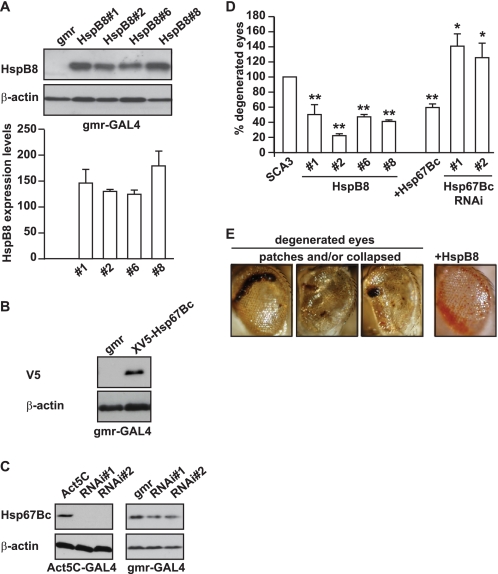
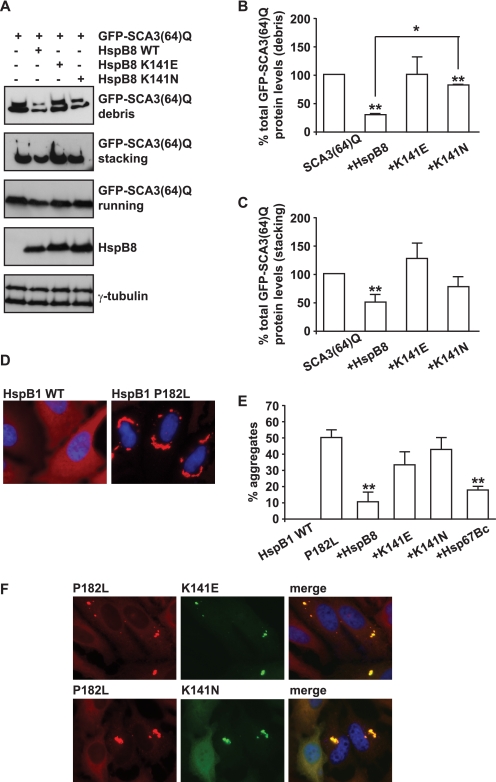
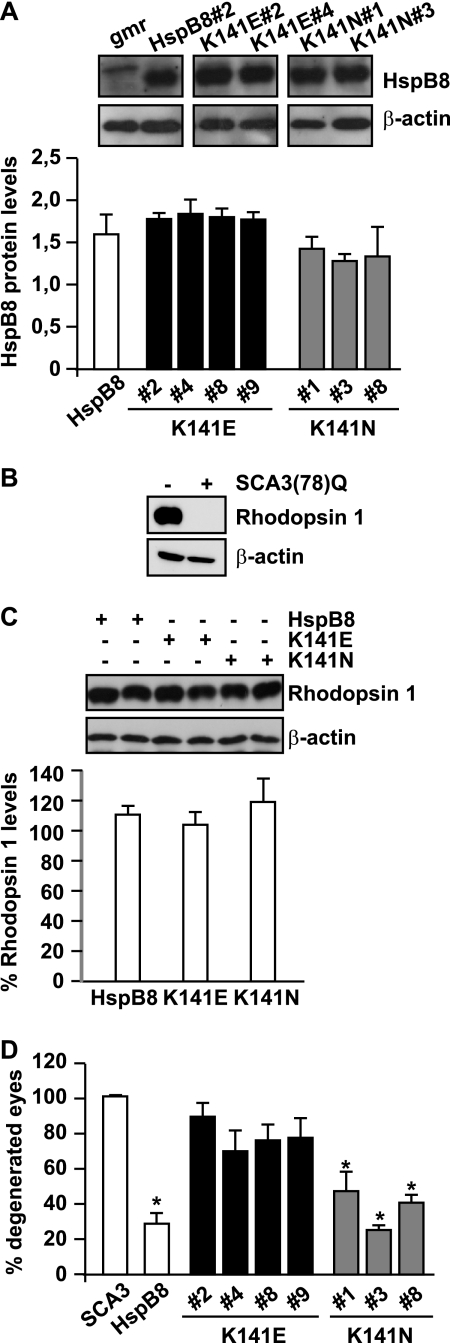
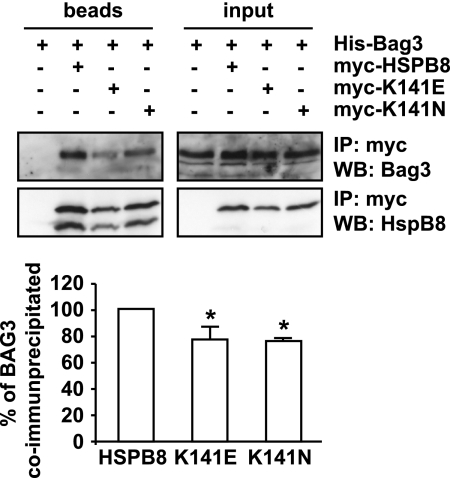
Protein aggregation is a hallmark of many neuronal disorders, including the polyglutamine disorder spinocerebellar ataxia 3 and peripheral neuropathies associated with the K141E and K141N mutations in the small heat shock protein HSPB8. In cells, HSPB8 cooperates with BAG3 to stimulate autophagy in an eIF2α-dependent manner and facilitates the clearance of aggregate-prone proteins (Carra, S., Seguin, S. J., Lambert, H., and Landry, J. (2008) J. Biol. Chem. 283, 1437-1444; Carra, S., Brunsting, J. F., Lambert, H., Landry, J., and Kampinga, H. H. (2009) J. Biol. Chem. 284, 5523-5532). Here, we first identified Drosophila melanogaster HSP67Bc (Dm-HSP67Bc) as the closest functional ortholog of human HSPB8 and demonstrated that, like human HSPB8, Dm-HSP67Bc induces autophagy via the eIF2α pathway. In vitro, both Dm-HSP67Bc and human HSPB8 protected against mutated ataxin-3-mediated toxicity and decreased the aggregation of a mutated form of HSPB1 (P182L-HSPB1) associated with peripheral neuropathy. Up-regulation of both Dm-HSP67Bc and human HSPB8 protected and down-regulation of endogenous Dm-HSP67Bc significantly worsened SCA3-mediated eye degeneration in flies. The K141E and K141N mutated forms of human HSPB8 that are associated with peripheral neuropathy were significantly less efficient than wild-type HSPB8 in decreasing the aggregation of both mutated ataxin 3 and P182L-HSPB1. Our current data further support the link between the HSPB8-BAG3 complex, autophagy, and folding diseases and demonstrate that impairment or loss of function of HSPB8 might accelerate the progression and/or severity of folding diseases.
Figures
Similar articles
-
The chaperone HSPB8 reduces the accumulation of truncated TDP-43 species in cells and protects against TDP-43-mediated toxicity.Hum Mol Genet. 2016 Sep 15;25(18):3908-3924. doi: 10.1093/hmg/ddw232. Epub 2016 Jul 27. Hum Mol Genet. 2016. PMID: 27466192 Free PMC article.
-
HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2{alpha} phosphorylation.J Biol Chem. 2009 Feb 27;284(9):5523-32. doi: 10.1074/jbc.M807440200. Epub 2008 Dec 29. J Biol Chem. 2009. PMID: 19114712
-
Autophagy induction by piplartine ameliorates axonal degeneration caused by mutant HSPB1 and HSPB8 in Charcot-Marie-Tooth type 2 neuropathies.Autophagy. 2025 May;21(5):1116-1143. doi: 10.1080/15548627.2024.2439649. Epub 2024 Dec 27. Autophagy. 2025. PMID: 39698979 Free PMC article.
-
THE FUNCTIONAL ROLE OF SMALL HEAT SHOCK PROTEIN Hsp67Bc IN DROSOPHILA MELANOGASTER.Tsitologiia. 2016;58(4):272-6. Tsitologiia. 2016. PMID: 30191693 Review. English, Russian.
-
HspB8 and Bag3: a new chaperone complex targeting misfolded proteins to macroautophagy.Autophagy. 2008 Feb;4(2):237-9. doi: 10.4161/auto.5407. Epub 2007 Dec 11. Autophagy. 2008. PMID: 18094623 Review.
Cited by
-
The α crystallin domain of small heat shock protein b8 (Hspb8) acts as survival and differentiation factor in adult hippocampal neurogenesis.J Neurosci. 2013 Mar 27;33(13):5785-96. doi: 10.1523/JNEUROSCI.6452-11.2013. J Neurosci. 2013. PMID: 23536091 Free PMC article.
-
Insights on Human Small Heat Shock Proteins and Their Alterations in Diseases.Front Mol Biosci. 2022 Feb 25;9:842149. doi: 10.3389/fmolb.2022.842149. eCollection 2022. Front Mol Biosci. 2022. PMID: 35281256 Free PMC article. Review.
-
Drosophila Hsp67Bc hot-spot variants alter muscle structure and function.Cell Mol Life Sci. 2018 Dec;75(23):4341-4356. doi: 10.1007/s00018-018-2875-z. Epub 2018 Jul 21. Cell Mol Life Sci. 2018. PMID: 30032358 Free PMC article.
-
The Regulation of the Small Heat Shock Protein B8 in Misfolding Protein Diseases Causing Motoneuronal and Muscle Cell Death.Front Neurosci. 2019 Aug 2;13:796. doi: 10.3389/fnins.2019.00796. eCollection 2019. Front Neurosci. 2019. PMID: 31427919 Free PMC article. Review.
-
The small heat shock protein B8 (HSPB8) modulates proliferation and migration of breast cancer cells.Oncotarget. 2017 Feb 7;8(6):10400-10415. doi: 10.18632/oncotarget.14422. Oncotarget. 2017. PMID: 28060751 Free PMC article.
References
-
- Cummings C. J., Zoghbi H. Y. (2000) Annu. Rev. Genomics Hum. Genet. 1, 281–328 - PubMed
-
- Ross C. A., Poirier M. A. (2004) Nat. Med. 10, (suppl.) S10–S17 - PubMed
-
- Fortun J., Go J. C., Li J., Amici S. A., Dunn W. A., Jr., Notterpek L. (2006) Neurobiol. Dis. 22, 153–164 - PubMed
-
- Fujita E., Kouroku Y., Isoai A., Kumagai H., Misutani A., Matsuda C., Hayashi Y. K., Momoi T. (2007) Hum. Mol. Genet. 16, 618–629 - PubMed
-
- Sharma M. C., Goebel H. H. (2005) Neurol. India 53, 273–279 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
