

Endogenous N-acetylaspartylglutamate (NAAG) inhibits synaptic plasticity/transmission in the amygdala in a mouse inflammatory pain model
- PMID: 20860833
- PMCID: PMC3152775
- DOI: 10.1186/1744-8069-6-60
Endogenous N-acetylaspartylglutamate (NAAG) inhibits synaptic plasticity/transmission in the amygdala in a mouse inflammatory pain model
Abstract
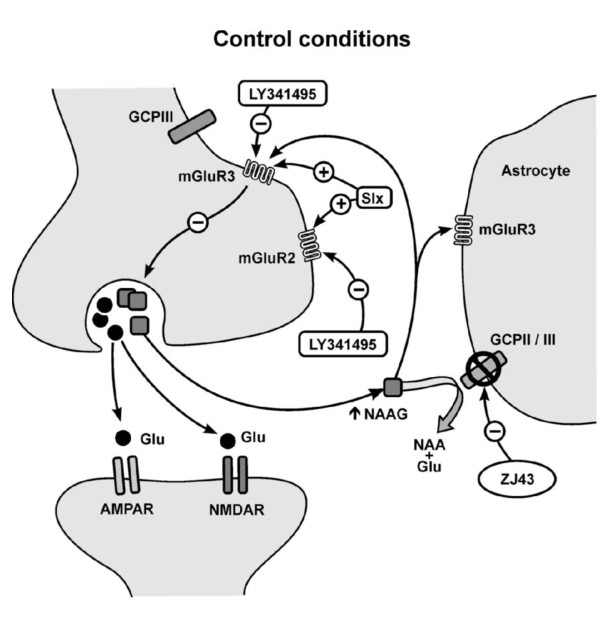
Background: The peptide neurotransmitter N-acetylaspartylglutamate (NAAG) is widely expressed throughout the vertebrate nervous system, including the pain processing neuraxis. Inhibitors of NAAG peptidases are analgesic in animal models of pain. However, the brain regions involved in NAAG's analgesic action have not been rigorously defined. Group II metabotropic glutamate receptors (mGluR2/3) play a role in pain processing in the laterocapsular part of the central nucleus of the amygdala (CeLC). Given the high concentration of NAAG in the amygdala and its activation of group II mGluRs (mGluR3 > mGluR2), this study was undertaken using the mouse formalin model of inflammatory pain to test the hypothesis that NAAG influences pain processing in the amygdala. Evoked excitatory postsynaptic currents (eEPSCs) were studied in neurons in the CeLC of mouse brain slices following stimulation of the spinoparabrachial amygdaloid afferents.
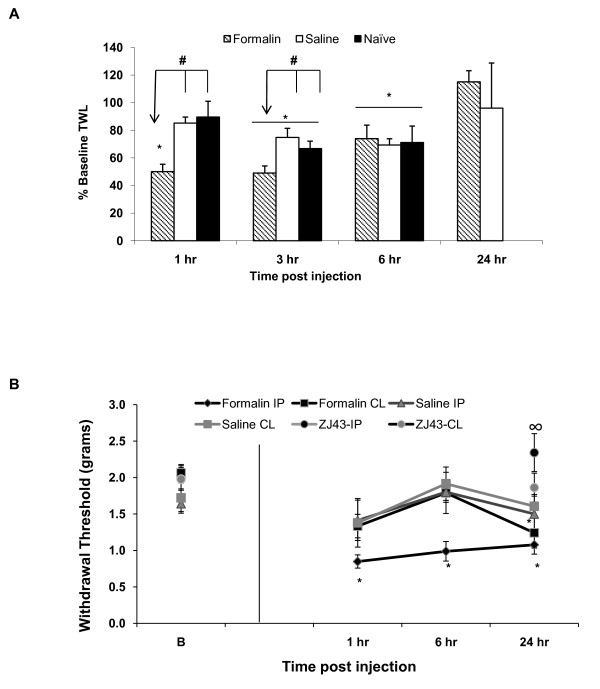
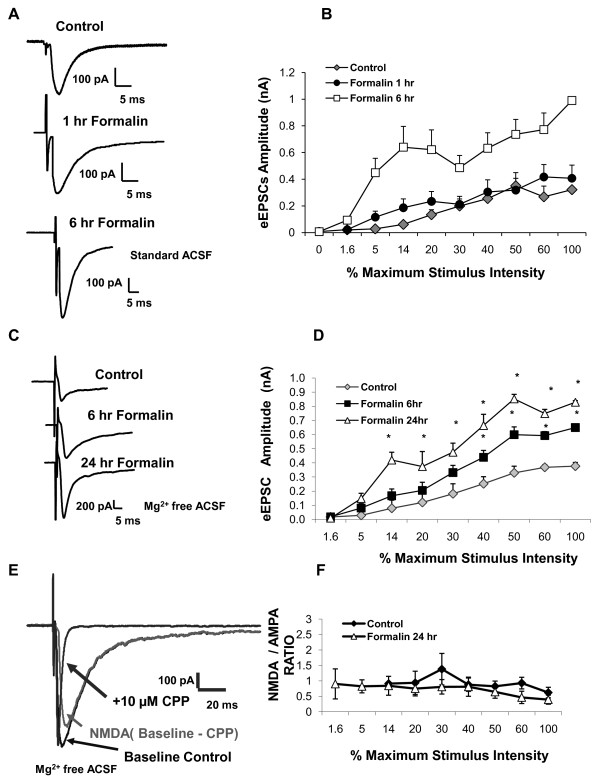
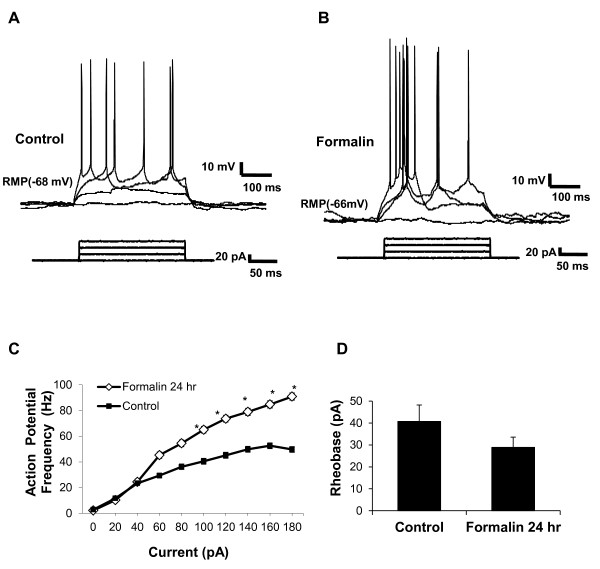
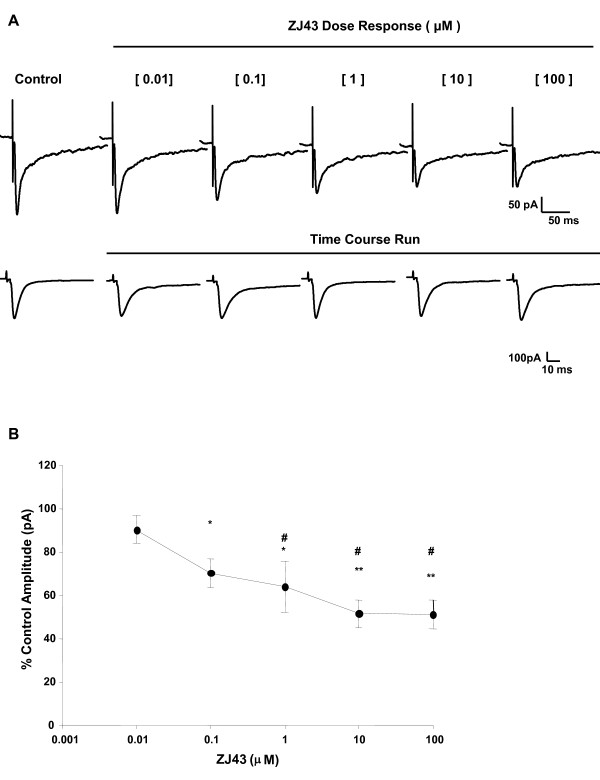
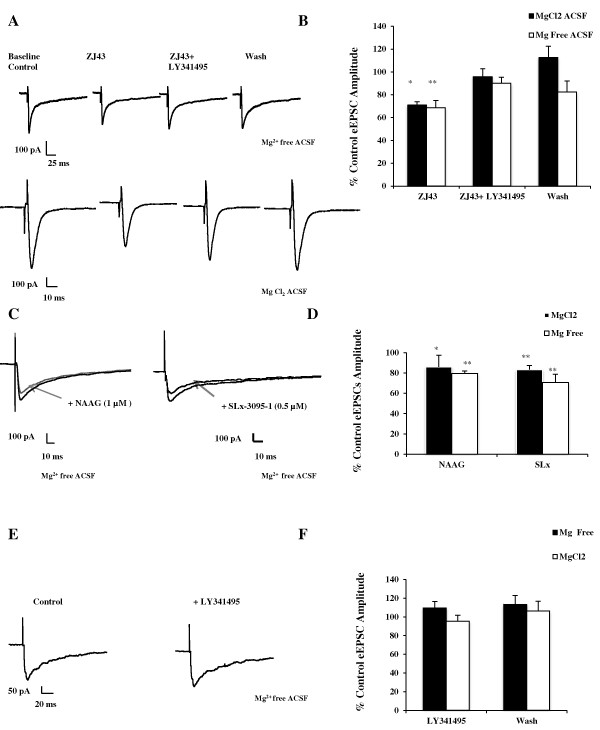
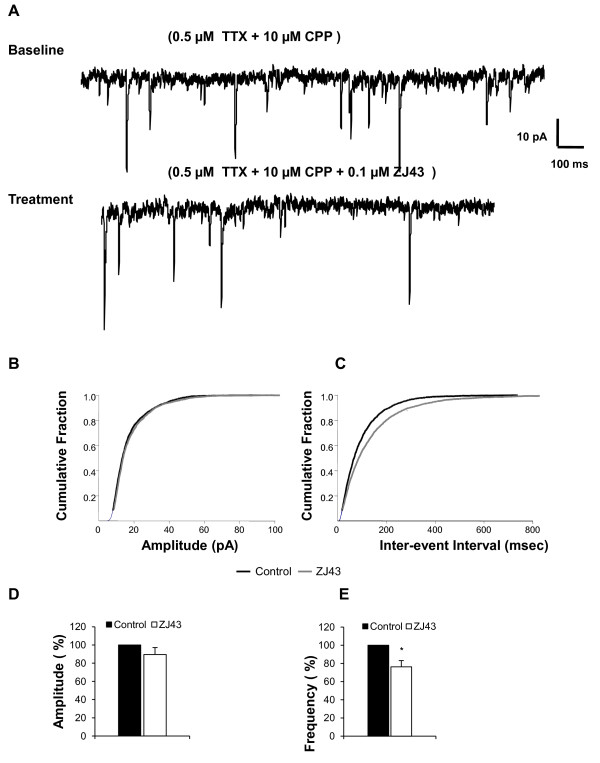
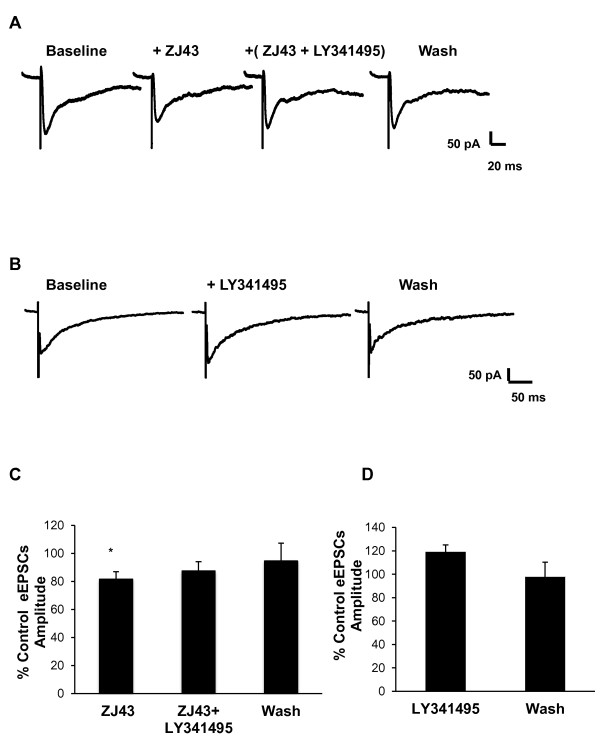
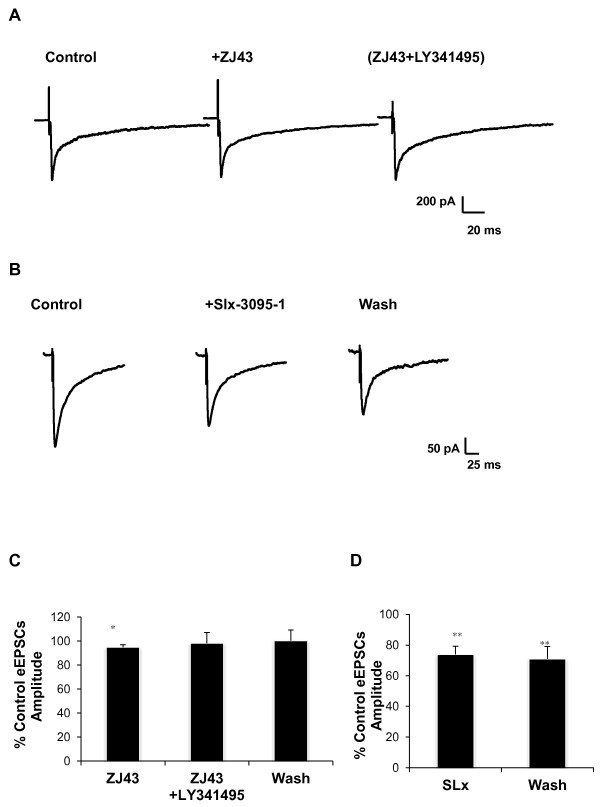
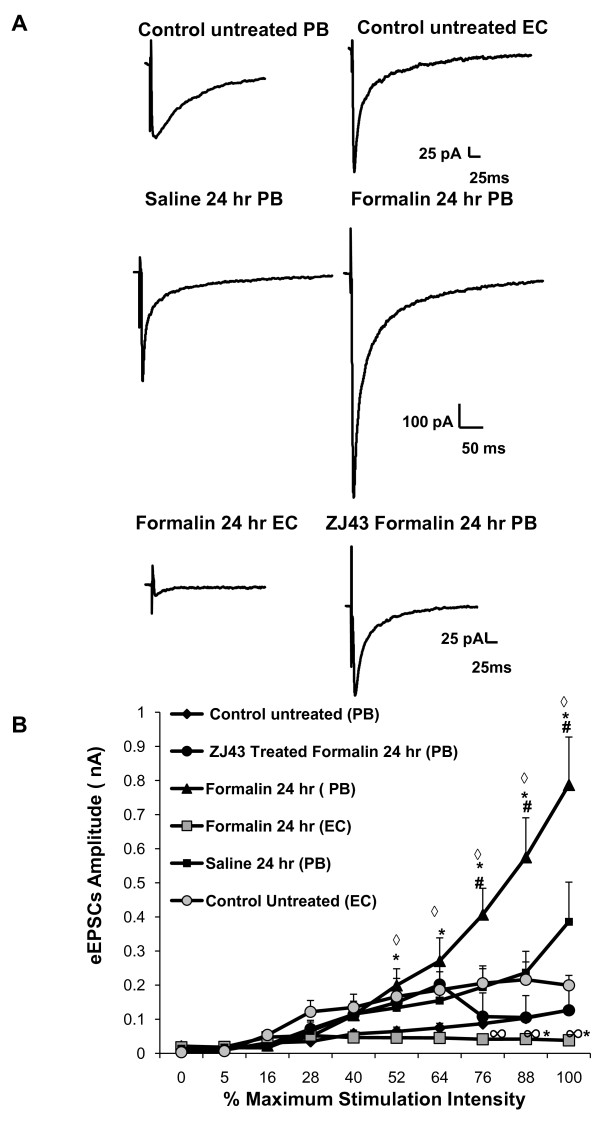
Results: Application of a NAAG peptidase inhibitor, ZJ43, dose dependently inhibited the amplitude of the eEPSCs by up to 50% in control CeLC demonstrating the role of NAAG in regulation of excitatory transmission at this synapse. A group II mGluR agonist (SLx-3095-1) similarly inhibited eEPSC amplitude by about 30%. Both effects were blocked by the group II mGluR antagonist LY341495. ZJ43 was much less effective than SLx in reducing eEPSCs 24 hours post inflammation suggesting an inflammation induced reduction in NAAG release or an increase in the ratio of mGluR2 to mGluR3 expression. Systemic injection of ZJ43 proximal to the time of inflammation blocked peripheral inflammation-induced increases in synaptic transmission of this pathway 24 hrs later and blocked the induction of mechanical allodynia that developed by this time point.
Conclusions: The main finding of this study is that NAAG and NAAG peptidase inhibition reduce excitatory neurotransmission and inflammation-induced plasticity at the spinoparabrachial synapse within the pain processing pathway of the central amygdaloid nucleus.
Figures
References
-
- Neale JH, Olszewski RT, Gehl LM, Wroblewska B, Bzdega T. The neurotransmitter N-acetylaspartylglutamate in models of pain, ALS, diabetic neuropathy, CNS injury and schizophrenia. Trends Pharmacol Sci. 2005;26:477–484. - PubMed
-
- Schweitzer C, Kratzeisen C, Adam G, Lundstrom K, Malherbe P, Ohresser S, Stadler H, Wichmann J, Woltering T, Mutel V. Characterization of [3H]-LY354740 binding to rat mGlu2 and mGlu3 receptors expressed in CHO cells using Semliki Forest virus vectors. Neuropharm. 2000;39:1700–1706. doi: 10.1016/S0028-3908(99)00265-8. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
