

NF-kappaB activation limits airway branching through inhibition of Sp1-mediated fibroblast growth factor-10 expression
- PMID: 20861353
- PMCID: PMC4399641
- DOI: 10.4049/jimmunol.1001857
NF-kappaB activation limits airway branching through inhibition of Sp1-mediated fibroblast growth factor-10 expression
Abstract
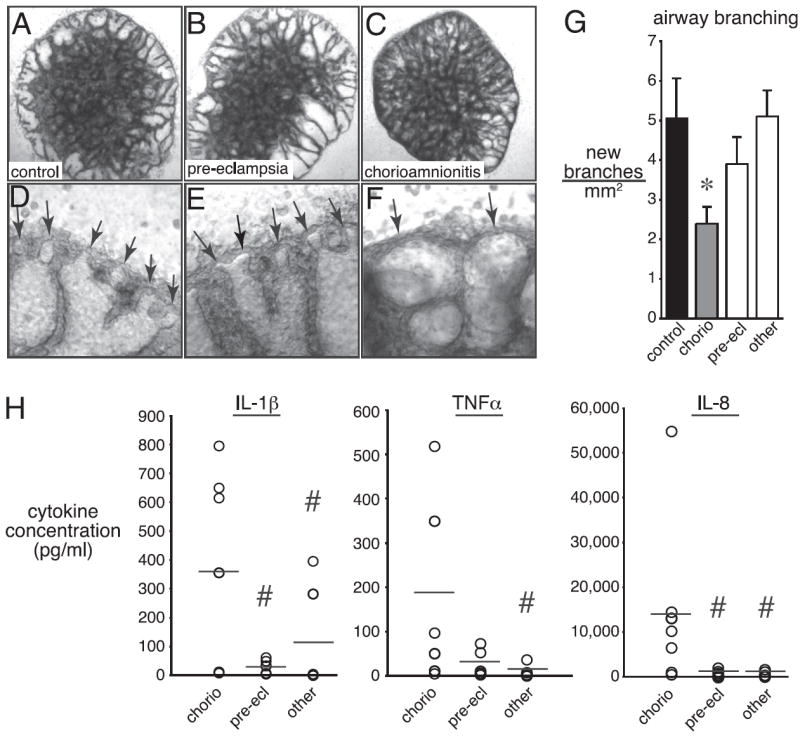
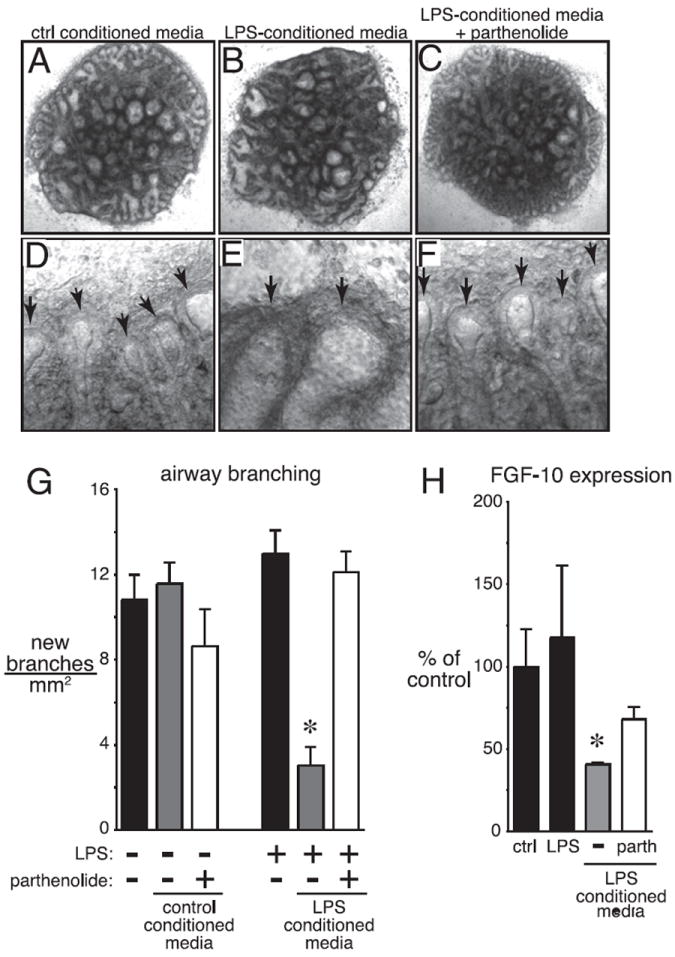
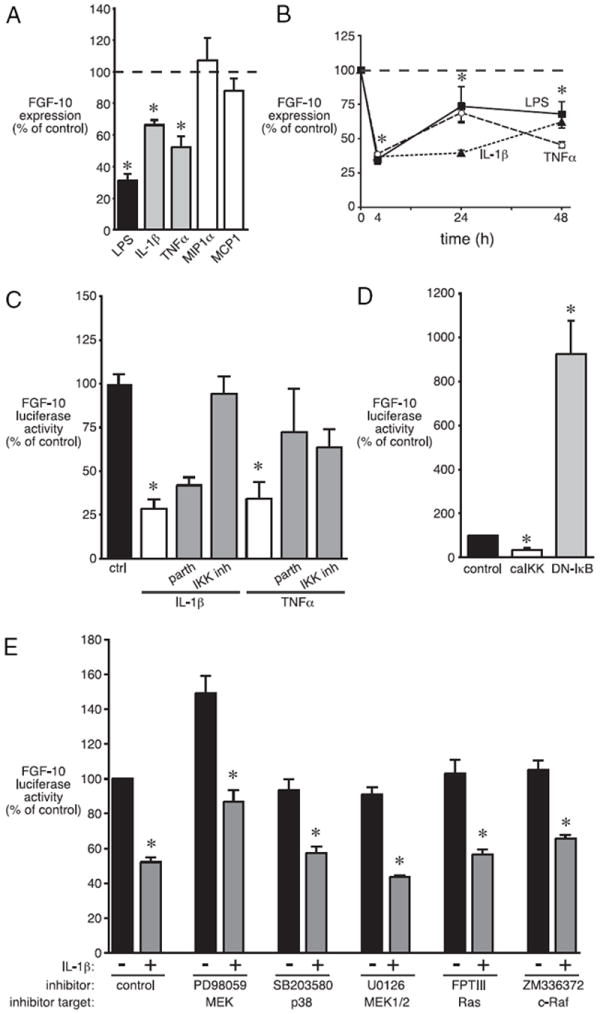
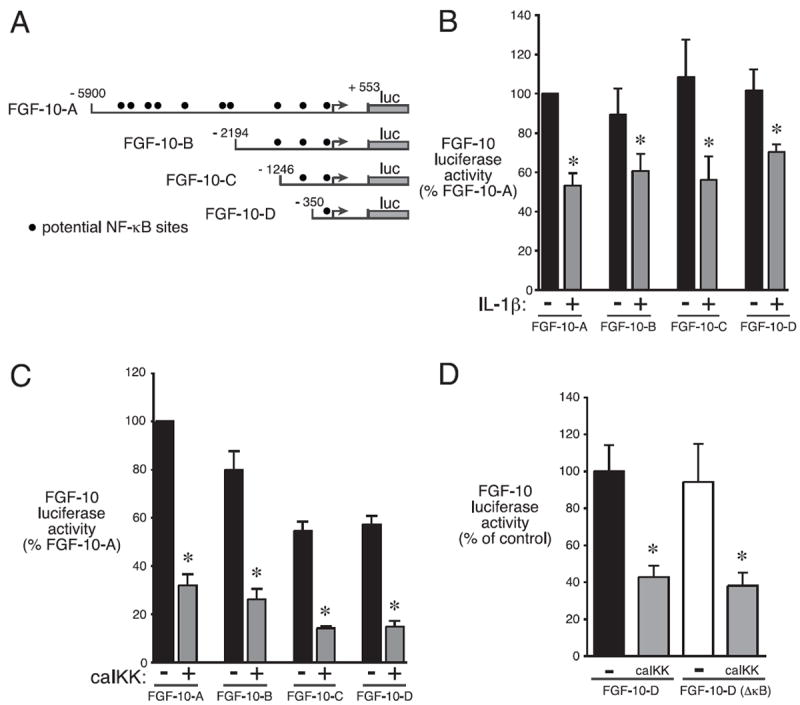
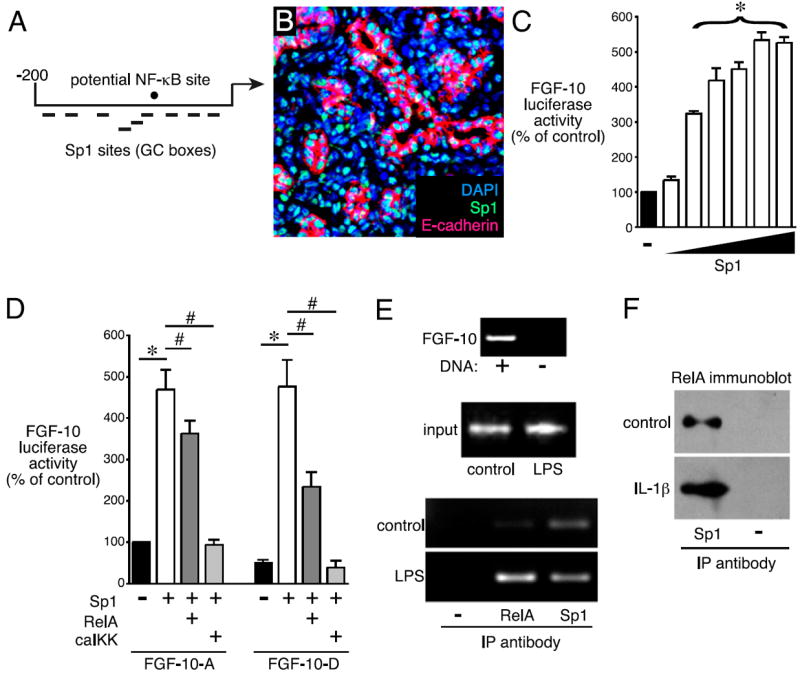
Bronchopulmonary dysplasia (BPD) is a frequent complication of preterm birth. This chronic lung disease results from arrested saccular airway development and is most common in infants exposed to inflammatory stimuli. In experimental models, inflammation inhibits expression of fibroblast growth factor-10 (FGF-10) and impairs epithelial-mesenchymal interactions during lung development; however, the mechanisms connecting inflammatory signaling with reduced growth factor expression are not yet understood. In this study we found that soluble inflammatory mediators present in tracheal fluid from preterm infants can prevent saccular airway branching. In addition, LPS treatment led to local production of mediators that inhibited airway branching and FGF-10 expression in LPS-resistant C.C3-Tlr4(Lpsd)/J fetal mouse lung explants. Both direct NF-κB activation and inflammatory cytokines (IL-1β and TNF-α) that activate NF-κB reduced FGF-10 expression, whereas chemokines that signal via other inflammatory pathways had no effect. Mutational analysis of the FGF-10 promoter failed to identify genetic elements required for direct NF-κB-mediated FGF-10 inhibition. Instead, NF-κB activation appeared to interfere with the normal stimulation of FGF-10 expression by Sp1. Chromatin immunoprecipitation and nuclear coimmunoprecipitation studies demonstrated that the RelA subunit of NF-κB and Sp1 physically interact at the FGF-10 promoter. These findings indicate that inflammatory signaling through NF-κB disrupts the normal expression of FGF-10 in fetal lung mesenchyme by interfering with the transcriptional machinery critical for lung morphogenesis.
Conflict of interest statement
The authors have no financial conflicts of interest.
Figures
Similar articles
-
Interactions between NF-κB and SP3 connect inflammatory signaling with reduced FGF-10 expression.J Biol Chem. 2013 May 24;288(21):15318-25. doi: 10.1074/jbc.M112.447318. Epub 2013 Apr 4. J Biol Chem. 2013. PMID: 23558680 Free PMC article.
-
NF-κB signaling in fetal lung macrophages disrupts airway morphogenesis.J Immunol. 2011 Sep 1;187(5):2740-7. doi: 10.4049/jimmunol.1101495. Epub 2011 Jul 20. J Immunol. 2011. PMID: 21775686 Free PMC article.
-
Lipopolysaccharide increases alveolar type II cell number in fetal mouse lungs through Toll-like receptor 4 and NF-kappaB.Am J Physiol Lung Cell Mol Physiol. 2004 Nov;287(5):L999-1006. doi: 10.1152/ajplung.00111.2004. Am J Physiol Lung Cell Mol Physiol. 2004. PMID: 15475494
-
Epithelial-Derived Inflammation Disrupts Elastin Assembly and Alters Saccular Stage Lung Development.Am J Pathol. 2016 Jul;186(7):1786-1800. doi: 10.1016/j.ajpath.2016.02.016. Epub 2016 May 12. Am J Pathol. 2016. PMID: 27181406 Free PMC article.
-
Copper metabolism domain-containing 1 represses the mediators involved in the terminal effector pathways of human labour and delivery.Mol Hum Reprod. 2016 Apr;22(4):299-310. doi: 10.1093/molehr/gav075. Epub 2016 Jan 4. Mol Hum Reprod. 2016. PMID: 26733542
Cited by
-
The effect of CSF-1 administration on lung maturation in a mouse model of neonatal hyperoxia exposure.Respir Res. 2014 Sep 6;15(1):110. doi: 10.1186/s12931-014-0110-5. Respir Res. 2014. PMID: 25192716 Free PMC article.
-
IL-1β and Inflammasome Activity Link Inflammation to Abnormal Fetal Airway Development.J Immunol. 2016 Apr 15;196(8):3411-20. doi: 10.4049/jimmunol.1500906. Epub 2016 Mar 7. J Immunol. 2016. PMID: 26951798 Free PMC article.
-
IKKβ Activation in the Fetal Lung Mesenchyme Alters Lung Vascular Development but Not Airway Morphogenesis.Am J Pathol. 2017 Dec;187(12):2635-2644. doi: 10.1016/j.ajpath.2017.08.013. Epub 2017 Sep 18. Am J Pathol. 2017. PMID: 28923684 Free PMC article.
-
LPS-induced chorioamnionitis and antenatal corticosteroids modulate Shh signaling in the ovine fetal lung.Am J Physiol Lung Cell Mol Physiol. 2012 Nov 1;303(9):L778-87. doi: 10.1152/ajplung.00280.2011. Epub 2012 Sep 7. Am J Physiol Lung Cell Mol Physiol. 2012. PMID: 22962010 Free PMC article.
-
Antenatal inflammation reduces expression of caveolin-1 and influences multiple signaling pathways in preterm fetal lungs.Am J Respir Cell Mol Biol. 2011 Nov;45(5):969-76. doi: 10.1165/rcmb.2010-0519OC. Epub 2011 May 11. Am J Respir Cell Mol Biol. 2011. PMID: 21562314 Free PMC article.
References
-
- Warburton D, Schwarz M, Tefft D, Flores-Delgado G, Anderson KD, Cardoso WV. The molecular basis of lung morphogenesis. Mech Dev. 2000;92:55–81. - PubMed
-
- Perl AK, Whitsett JA. Molecular mechanisms controlling lung morphogenesis. Clin Genet. 1999;56:14–27. - PubMed
-
- Hogan BL, Yingling JM. Epithelial/mesenchymal interactions and branching morphogenesis of the lung. Curr Opin Genet Dev. 1998;8:481–486. - PubMed
-
- Weaver M, Batts L, Hogan BL. Tissue interactions pattern the mesenchyme of the embryonic mouse lung. Dev Biol. 2003;258:169–184. - PubMed
-
- Weaver M, Dunn NR, Hogan BL. Bmp4 and Fgf10 play opposing roles during lung bud morphogenesis. Development. 2000;127:2695–2704. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P60 DK020593/DK/NIDDK NIH HHS/United States
- DK58404/DK/NIDDK NIH HHS/United States
- R01 AI079253/AI/NIAID NIH HHS/United States
- P30 DK058404/DK/NIDDK NIH HHS/United States
- P30 HD015052/HD/NICHD NIH HHS/United States
- HL-097195/HL/NHLBI NIH HHS/United States
- EY08126/EY/NEI NIH HHS/United States
- P30 EY008126/EY/NEI NIH HHS/United States
- R01 HL097195/HL/NHLBI NIH HHS/United States
- P30 CA068485/CA/NCI NIH HHS/United States
- CA68485/CA/NCI NIH HHS/United States
- AI-079253/AI/NIAID NIH HHS/United States
- HD15052/HD/NICHD NIH HHS/United States
- DK20593/DK/NIDDK NIH HHS/United States
- U24 DK059637/DK/NIDDK NIH HHS/United States
- HL-086324/HL/NHLBI NIH HHS/United States
- R01 HL086324/HL/NHLBI NIH HHS/United States
- DK59637/DK/NIDDK NIH HHS/United States
- P30 DK020593/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
