

Chronic dysfunction of astrocytic inwardly rectifying K+ channels specific to the neocortical epileptic focus after fluid percussion injury in the rat
- PMID: 20861444
- PMCID: PMC3007644
- DOI: 10.1152/jn.00398.2010
Chronic dysfunction of astrocytic inwardly rectifying K+ channels specific to the neocortical epileptic focus after fluid percussion injury in the rat
Abstract
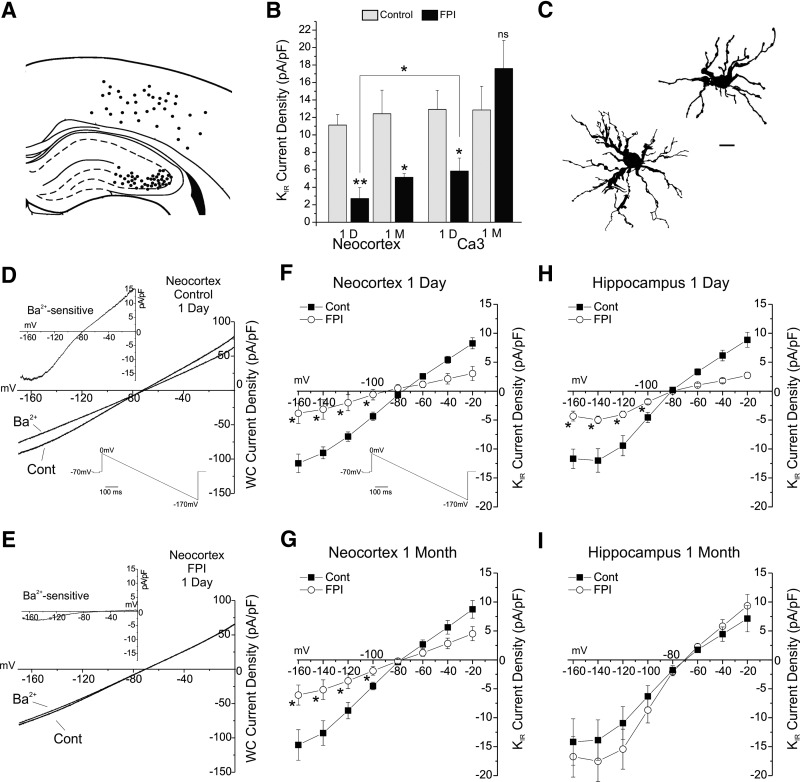
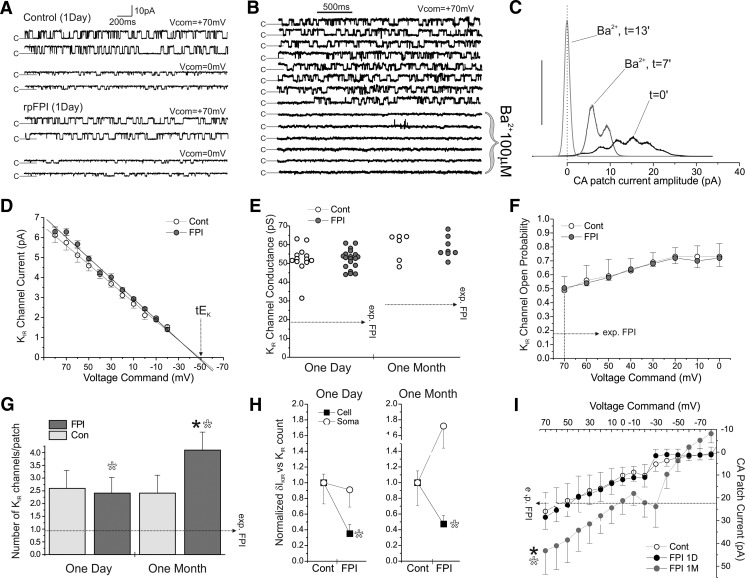
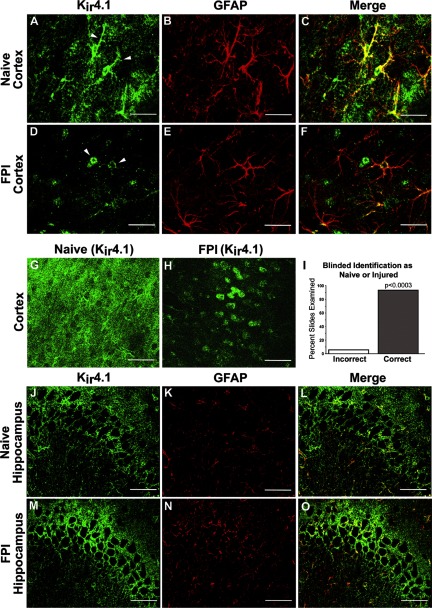
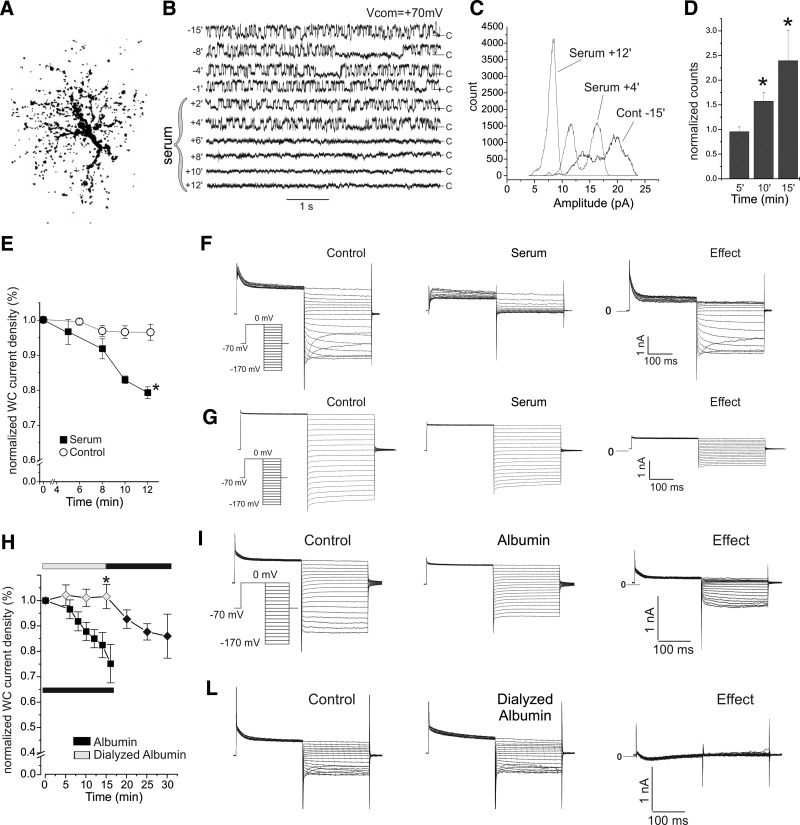
Astrocytic inwardly rectifying K(+) currents (I(KIR)) have an important role in extracellular K(+) homeostasis, which influences neuronal excitability, and serum extravasation has been linked to impaired K(IR)-mediated K(+) buffering and chronic hyperexcitability. Head injury induces acute impairment in astroglial membrane I(KIR) and impaired K(+) buffering in the rat hippocampus, but chronic spontaneous seizures appear in the perilesional neocortex--not the hippocampus--in the early weeks to months after injury. Thus we examined astrocytic K(IR) channel pathophysiology in both neocortex and hippocampus after rostral parasaggital fluid percussion injury (rpFPI). rpFPI induced greater acute serum extravasation and metabolic impairment in the perilesional neocortex than in the underlying hippocampus, and in situ whole cell recordings showed a greater acute loss of astrocytic I(KIR) in neocortex than hippocampus. I(KIR) loss persisted through 1 mo after injury only in the neocortical epileptic focus, but fully recovered in the hippocampus that did not generate chronic seizures. Neocortical cell-attached recordings showed no loss or an increase of I(KIR) in astrocytic somata. Confocal imaging showed depletion of KIR4.1 immunoreactivity especially in processes--not somata--of neocortical astrocytes, whereas hippocampal astrocytes appeared normal. In naïve animals, intracortical infusion of serum, devoid of coagulation-mediating thrombin activity, reproduces the effects of rpFPI both in vivo and at the cellular level. In vivo serum infusion induces partial seizures similar to those induced by rpFPI, whereas bath-applied serum, but not dialyzed albumin, rapidly silenced astrocytic K(IR) membrane currents in whole cell and cell-attached patch-clamp recordings in situ. Thus both acute impairment in astrocytic I(KIR) and chronic spontaneous seizures typical of rpFPI are reproduced by serum extravasation, whereas the chronic impairment in astroglial I(KIR) is specific to the neocortex that develops the epileptic focus.
Figures
References
-
- Abrahám H, Losonczy A, Czéh G, Lázár G. Rapid activation of microglial cells by hypoxia, kainic acid, and potassium ions in slice preparations of the rat hippocampus. Brain Res 906: 115–126, 2001 - PubMed
-
- Albensi BC, Janigro D. Traumatic brain injury and its effects on synaptic plasticity. Brain Inj 17: 653–663, 2003 - PubMed
-
- Alexander JS, Patton WF, Christman BW, Cuiper LL, Haselton FR. Platelet-derived lysophosphatidic acid decreases endothelial permeability in vitro Am J Physiol Heart Circ Physiol 274: H115–H122, 1998 - PubMed
-
- Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. N Engl J Med 338: 20–24, 1998 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources