

The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism
- PMID: 20861672
- PMCID: PMC3047615
- DOI: 10.4161/cc.9.17.12721
The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism
Abstract
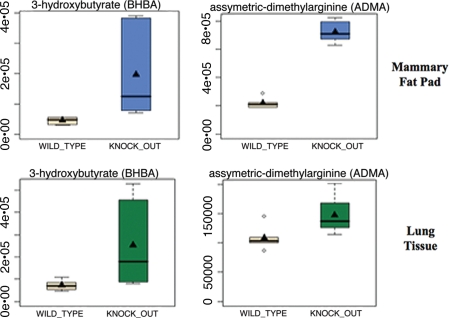
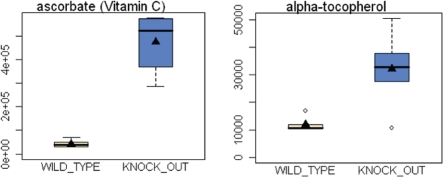
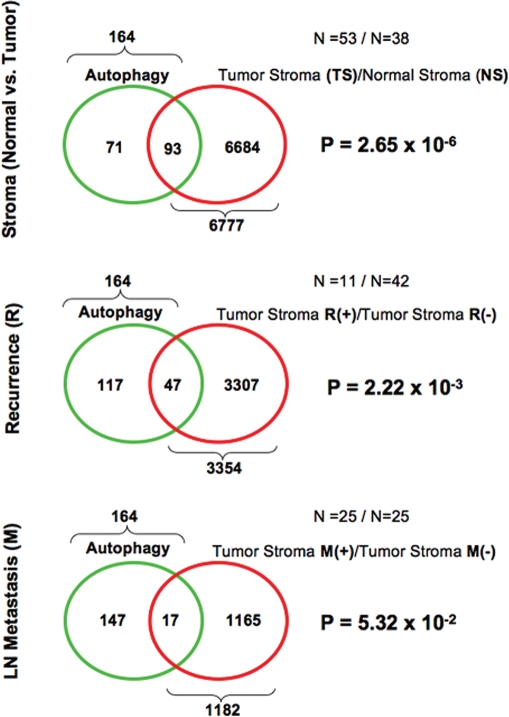
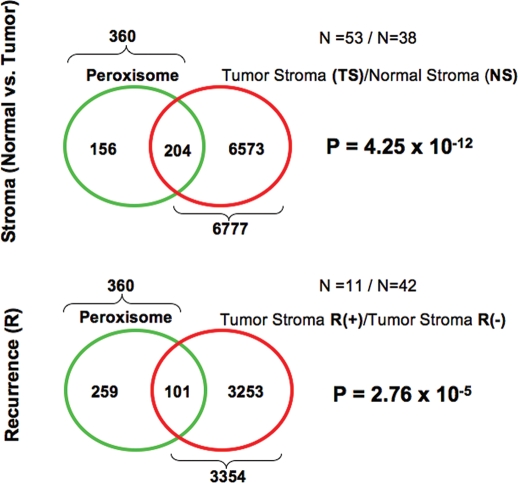
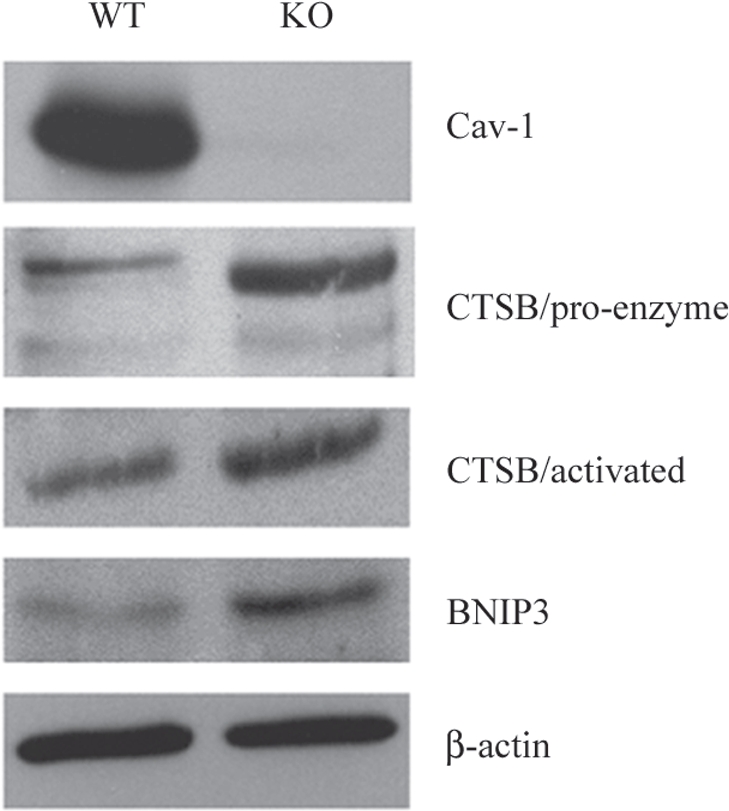
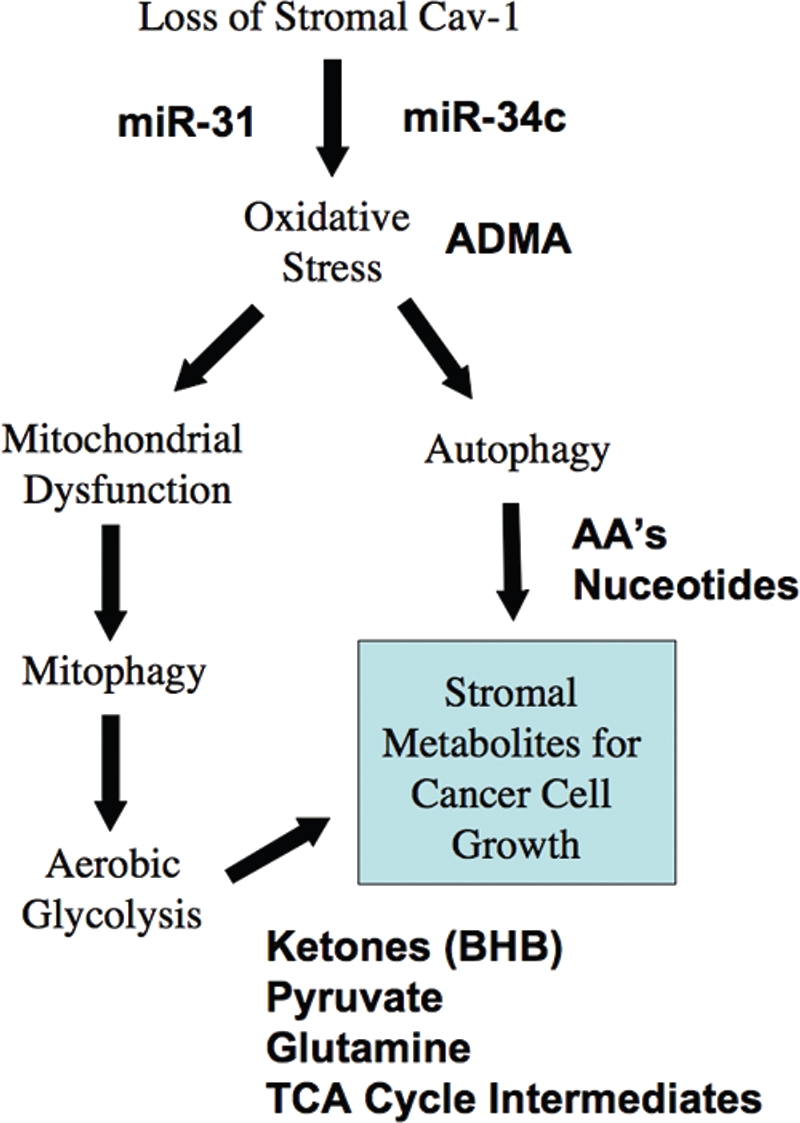
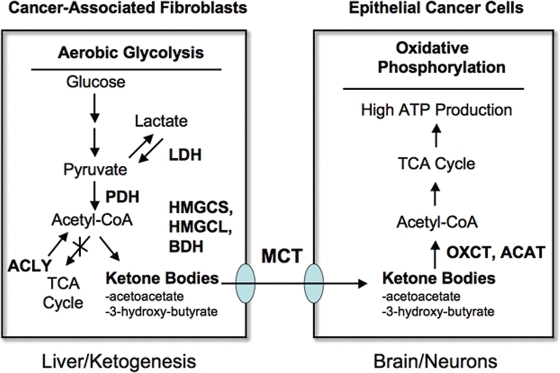
A loss of stromal Cav-1 in the tumor fibroblast compartment is associated with early tumor recurrence, lymph-node metastasis, and tamoxifen-resistance, resulting in poor clinical outcome in breast cancer patients. Here, we have used Cav-1 (-/-) null mice as a pre-clinical model for this "lethal tumor micro-environment." Metabolic profiling of Cav-1 (-/-) mammary fat pads revealed the upregulation of numerous metabolites (nearly 100), indicative of a major catabolic phenotype. Our results are consistent with the induction of oxidative stress, mitochondrial dysfunction, and autophagy/mitophagy. The two most prominent metabolites that emerged from this analysis were ADMA (asymmetric dimethyl arginine) and BHB (beta-hydroxybutyrate; a ketone body), which are markers of oxidative stress and mitochondrial dysfunction, respectively. Transcriptional profiling of Cav-1 (-/-) stromal cells and human tumor stroma from breast cancer patients directly supported an association with oxidative stress, mitochondrial dysfunction, and autophagy/mitophagy, as well as ADMA and ketone production. MircoRNA profiling of Cav-1 (-/-) stromal cells revealed the upregulation of two key cancer-related miR's, namely miR-31 and miR-34c. Consistent with our metabolic findings, these miR's are associated with oxidative stress (miR-34c) or activation of the hypoxic response/HIF1a (miR-31), which is sufficient to drive authophagy/mitophagy. Thus, via an unbiased comprehensive analysis of a lethal tumor micro-environment, we have identified a number of candidate biomarkers (ADMA, ketones, and miR-31/34c) that could be used to identify high-risk cancer patients at diagnosis, for treatment stratification and/or for evaluating therapeutic efficacy during anti-cancer therapy. We propose that the levels of these key biomarkers (ADMA, ketones/BHB, miR-31, and miR-34c) could be (1) assayed using serum or plasma from cancer patients, or (2) performed directly on excised tumor tissue. Importantly, induction of oxidative stress and autophagy/mitophagy in the tumor stromal compartment provides a means by which epithelial cancer cells can directly "feed off" of stromal-derived essential nutrients, chemical building blocks (amino acids, nucleotides), and energy-rich metabolites (glutamine, pyruvate, ketones/BHB), driving tumor progression and metastasis. Essentially, aggressive cancer cells are "eating" the cancer-associated fibroblasts via autophagy/mitophagy in the tumor micro-environment. Lastly, we discuss that this "Autophagic Tumor Stroma Model of Cancer Metabolism" provides a viable solution to the "Autophagy Paradox" in cancer etiology and chemo-therapy.
Figures


References
-
- Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA075503/CA/NCI NIH HHS/United States
- R01 CA098779/CA/NCI NIH HHS/United States
- R01-CA-120876/CA/NCI NIH HHS/United States
- R01 CA120876/CA/NCI NIH HHS/United States
- R01-CA-70896/CA/NCI NIH HHS/United States
- R01-CA-098779/CA/NCI NIH HHS/United States
- R01-CA-86072/CA/NCI NIH HHS/United States
- R01-AR-055660/AR/NIAMS NIH HHS/United States
- R01-CA-080250/CA/NCI NIH HHS/United States
- R01 CA070896/CA/NCI NIH HHS/United States
- R01 CA107382/CA/NCI NIH HHS/United States
- P30 CA056036/CA/NCI NIH HHS/United States
- P30-CA-56036/CA/NCI NIH HHS/United States
- R01-CA-107382/CA/NCI NIH HHS/United States
- R01 AR055660/AR/NIAMS NIH HHS/United States
- R01-CA-75503/CA/NCI NIH HHS/United States
- R01 CA080250/CA/NCI NIH HHS/United States
- R01 CA086072/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical