

Mice doubly-deficient in lysosomal hexosaminidase A and neuraminidase 4 show epileptic crises and rapid neuronal loss
- PMID: 20862357
- PMCID: PMC2940724
- DOI: 10.1371/journal.pgen.1001118
Mice doubly-deficient in lysosomal hexosaminidase A and neuraminidase 4 show epileptic crises and rapid neuronal loss
Abstract
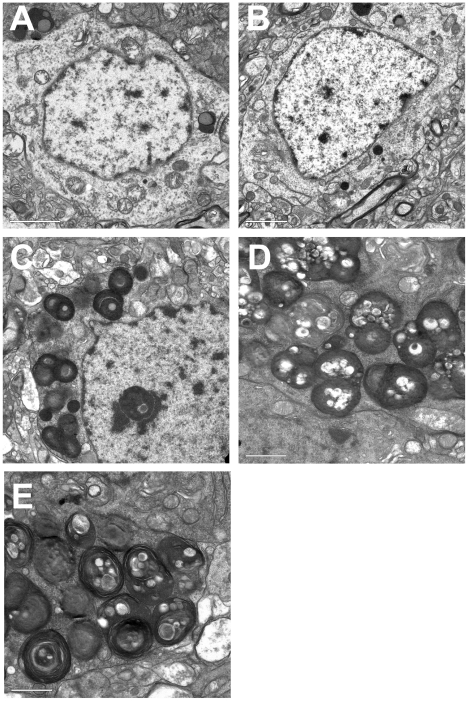
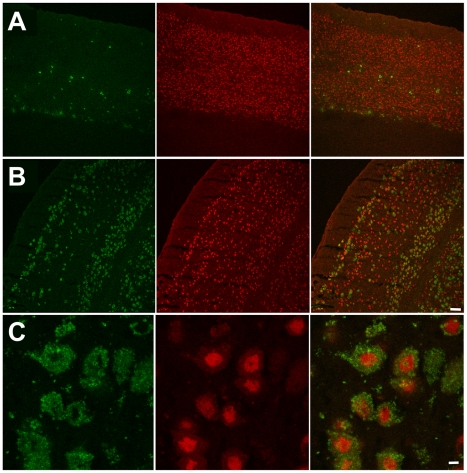
Tay-Sachs disease is a severe lysosomal disorder caused by mutations in the HexA gene coding for the α-subunit of lysosomal β-hexosaminidase A, which converts G(M2) to G(M3) ganglioside. Hexa(-/-) mice, depleted of β-hexosaminidase A, remain asymptomatic to 1 year of age, because they catabolise G(M2) ganglioside via a lysosomal sialidase into glycolipid G(A2), which is further processed by β-hexosaminidase B to lactosyl-ceramide, thereby bypassing the β-hexosaminidase A defect. Since this bypass is not effective in humans, infantile Tay-Sachs disease is fatal in the first years of life. Previously, we identified a novel ganglioside metabolizing sialidase, Neu4, abundantly expressed in mouse brain neurons. Now we demonstrate that mice with targeted disruption of both Neu4 and Hexa genes (Neu4(-/-);Hexa(-/-)) show epileptic seizures with 40% penetrance correlating with polyspike discharges on the cortical electrodes of the electroencephalogram. Single knockout Hexa(-/-) or Neu4(-/-) siblings do not show such symptoms. Further, double-knockout but not single-knockout mice have multiple degenerating neurons in the cortex and hippocampus and multiple layers of cortical neurons accumulating G(M2) ganglioside. Together, our data suggest that the Neu4 block exacerbates the disease in Hexa(-/-) mice, indicating that Neu4 is a modifier gene in the mouse model of Tay-Sachs disease, reducing the disease severity through the metabolic bypass. However, while disease severity in the double mutant is increased, it is not profound suggesting that Neu4 is not the only sialidase contributing to the metabolic bypass in Hexa(-/-) mice.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures





Similar articles
-
Mice deficient in Neu4 sialidase exhibit abnormal ganglioside catabolism and lysosomal storage.Hum Mol Genet. 2008 Jun 1;17(11):1556-68. doi: 10.1093/hmg/ddn043. Epub 2008 Feb 11. Hum Mol Genet. 2008. PMID: 18270209
-
Murine Sialidase Neu3 facilitates GM2 degradation and bypass in mouse model of Tay-Sachs disease.Exp Neurol. 2018 Jan;299(Pt A):26-41. doi: 10.1016/j.expneurol.2017.09.012. Epub 2017 Sep 30. Exp Neurol. 2018. PMID: 28974375
-
GM2 ganglioside accumulation causes neuroinflammation and behavioral alterations in a mouse model of early onset Tay-Sachs disease.J Neuroinflammation. 2020 Sep 20;17(1):277. doi: 10.1186/s12974-020-01947-6. J Neuroinflammation. 2020. PMID: 32951593 Free PMC article.
-
The beta-hexosaminidase deficiency disorders: development of a clinical paradigm in the mouse.Ann Med. 1997 Dec;29(6):557-61. doi: 10.3109/07853899709007482. Ann Med. 1997. PMID: 9562524 Review.
-
New Approaches to Tay-Sachs Disease Therapy.Front Physiol. 2018 Nov 20;9:1663. doi: 10.3389/fphys.2018.01663. eCollection 2018. Front Physiol. 2018. PMID: 30524313 Free PMC article. Review.
Cited by
-
Novel Vector Design and Hexosaminidase Variant Enabling Self-Complementary Adeno-Associated Virus for the Treatment of Tay-Sachs Disease.Hum Gene Ther. 2016 Jul;27(7):509-21. doi: 10.1089/hum.2016.013. Hum Gene Ther. 2016. PMID: 27197548 Free PMC article.
-
Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model.Brain. 2015 Feb;138(Pt 2):336-55. doi: 10.1093/brain/awu355. Epub 2015 Jan 6. Brain. 2015. PMID: 25567323 Free PMC article.
-
Substrate Reduction Therapy for Sandhoff Disease through Inhibition of Glucosylceramide Synthase Activity.Mol Ther. 2019 Aug 7;27(8):1495-1506. doi: 10.1016/j.ymthe.2019.05.018. Epub 2019 Jun 4. Mol Ther. 2019. PMID: 31208914 Free PMC article.
-
Neuraminidase-1 contributes significantly to the degradation of neuronal B-series gangliosides but not to the bypass of the catabolic block in Tay-Sachs mouse models.Mol Genet Metab Rep. 2015 Aug 15;4:72-82. doi: 10.1016/j.ymgmr.2015.07.004. eCollection 2015 Sep. Mol Genet Metab Rep. 2015. PMID: 26937414 Free PMC article.
-
Characterization of a phenotypically severe animal model for human AB-Variant GM2 gangliosidosis.Front Mol Neurosci. 2023 Nov 30;16:1242814. doi: 10.3389/fnmol.2023.1242814. eCollection 2023. Front Mol Neurosci. 2023. PMID: 38098938 Free PMC article.
References
-
- Gravel RA, Kaback MM, Proia RL, Sandhoff K, Suzuki K, et al. The GM2 Gangliosidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. New York: McGraw-Hill; 2001. pp. 3827–3876.
-
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. - PubMed
-
- Kaback MM, Rimoin DL, O'Brien JS. New York: Alan R. Liss (pub.); 1977. Tay-Sachs Disease: Screening and Prevention.
-
- Andermann E, Scriver CR, Wolfe LS, Dansky L, Andermann F. Genetic variants of Tay-Sachs disease and Sandhoff's disease in French-Canadians, juvenile Tay-Sachs disease in Lebanese Canadians, and a Tay-Sachs screening program in the French-Canadian population. In: Kaback MM, Rimoin DL, O'Brien JS, editors. Tay-Sachs Disease: Screening and Prevention. New York: Alan R. Liss (pub.); 1977. pp. 161–168. - PubMed
-
- Sango K, Yamanaka S, Hoffmann A, Okuda Y, Grinberg A, et al. Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat Genet. 1995;11:170–176. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
