

Integrated post-experiment monoisotopic mass refinement: an integrated approach to accurately assign monoisotopic precursor masses to tandem mass spectrometric data
- PMID: 20863060
- PMCID: PMC3019303
- DOI: 10.1021/ac101388b
Integrated post-experiment monoisotopic mass refinement: an integrated approach to accurately assign monoisotopic precursor masses to tandem mass spectrometric data
Abstract
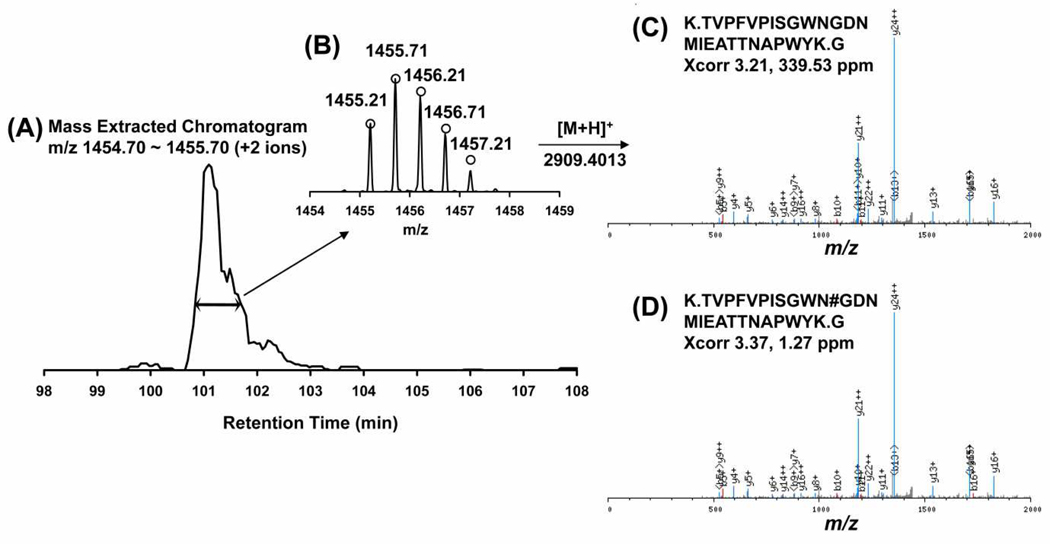
Accurate assignment of monoisotopic precursor masses to tandem mass spectrometric (MS/MS) data is a fundamental and critically important step for successful peptide identifications in mass spectrometry based proteomics. Here we describe an integrated approach that combines three previously reported methods of treating MS/MS data for precursor mass refinement. This combined method, "integrated post-experiment monoisotopic mass refinement" (iPE-MMR), integrates steps (1) generation of refined MS/MS data by DeconMSn; (2) additional refinement of the resultant MS/MS data by a modified version of PE-MMR; and (3) elimination of systematic errors of precursor masses using DtaRefinery. iPE-MMR is the first method that utilizes all MS information from multiple MS scans of a precursor ion including multiple charge states, in an MS scan, to determine precursor mass. With the combination of these methods, iPE-MMR increases sensitivity in peptide identification and provides increased accuracy when applied to complex high-throughput proteomics data.
Figures






References
-
- Shin B, Jung H-J, Hyung S-W, Kim H, Lee D, Lee C, Yu M-H, Lee S-W. Mol. Cell. Proteomics. 2008;7:1124–1134. - PubMed
-
- Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. Nat. Biotechnol. 2006;24:1285–1292. - PubMed
-
- Bakalarski CE, Haas W, Dephoure NE, Gygi SP. Anal. Bioanal. Chem. 2007;389:1409–1419. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
