

Mechanisms and cell signaling in alcoholic liver disease
- PMID: 20868231
- PMCID: PMC3755482
- DOI: 10.1515/BC.2010.137
Mechanisms and cell signaling in alcoholic liver disease
Abstract
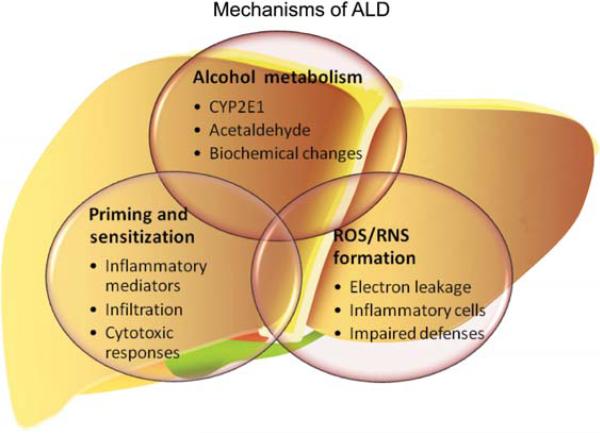
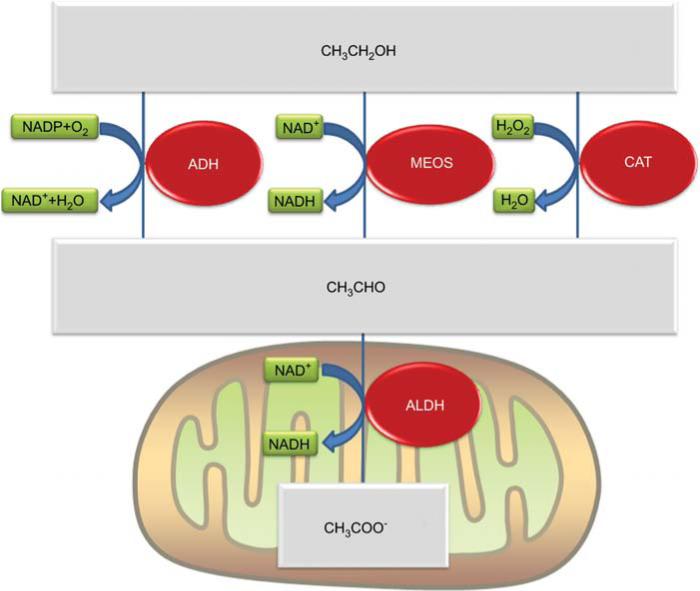
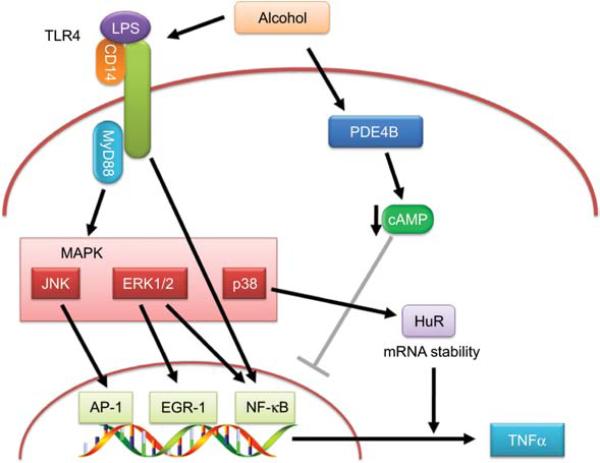
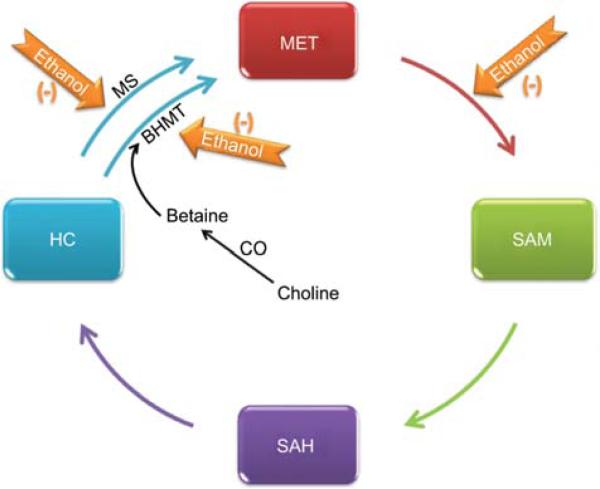
Alcoholic liver disease (ALD) remains a major cause of morbidity and mortality worldwide. For example, the Veterans Administration Cooperative Studies reported that patients with cirrhosis and superimposed alcoholic hepatitis had a 4-year mortality of >60%. The poor prognosis of ALD implies that preventing disease progression would be more effective than treating end-stage liver disease. An obvious avenue of prevention would be to remove the damaging agent; however, the infamously high rate of recidivism in alcoholics makes maintaining abstinence a difficult treatment goal to prevent ALD. Indeed, although the progression of ALD is well-characterized, there is no universally accepted therapy available to halt or reverse this process in humans. With better understanding of the mechanism(s) and risk factors that mediate the initiation and progression of ALD, rational targeted therapy can be developed to treat or prevent ALD. The purpose of this review is to summarize the established and proposed mechanisms by which chronic alcohol abuse damages the liver and to highlight key signaling events known or hypothesized to mediate these effects.
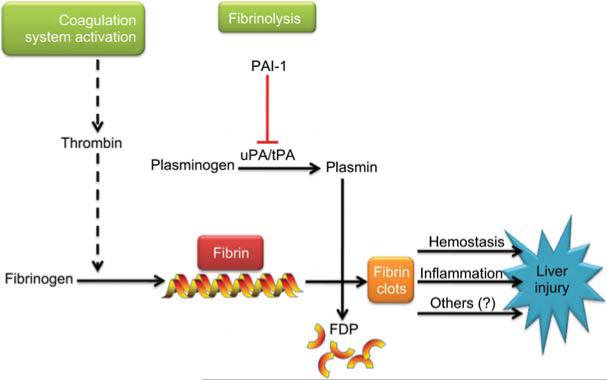
Figures
References
-
- Adachi Y, Bradford BU, Gao W, Bojes HK, Thurman RG. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology. 1994;20:453–460. - PubMed
-
- Allen RG, Tresini M. Oxidative stress and gene regulation. Free Radic. Biol. Med. 2000;28:463–499. - PubMed
-
- Arteel GE. Oxidants and antioxidants in alcohol-induced liver disease. Gastroenterology. 2003;124:778–790. - PubMed
-
- Arteel GE, Raleigh JA, Bradford BU, Thurman RG. Acute alcohol produces hypoxia directly in rat liver tissue in vivo: role of Kupffer cells. Am. J. Physiol. 1996;271:G494–G500. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical