

Dietary hyperglycemia, glycemic index and metabolic retinal diseases
- PMID: 20868767
- PMCID: PMC4005015
- DOI: 10.1016/j.preteyeres.2010.09.001
Dietary hyperglycemia, glycemic index and metabolic retinal diseases
Abstract
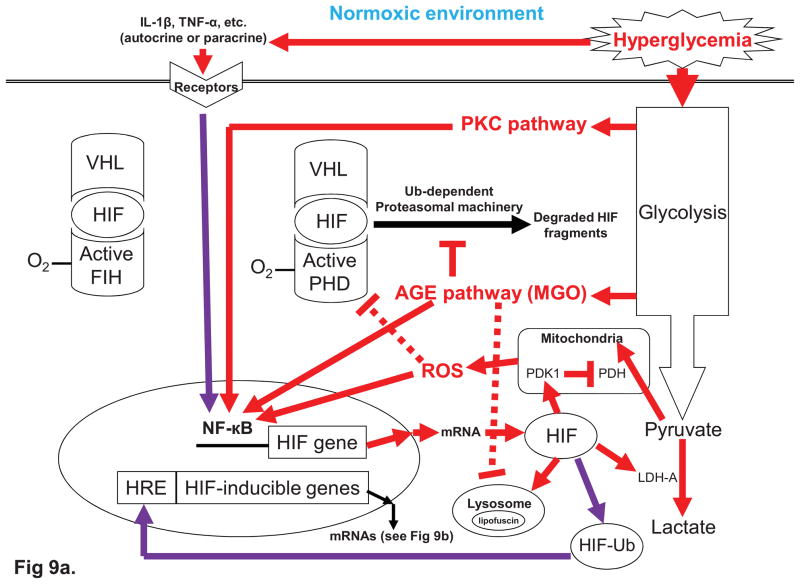
The glycemic index (GI) indicates how fast blood glucose is raised after consuming a carbohydrate-containing food. Human metabolic studies indicate that GI is related to patho-physiological responses after meals. Compared with a low-GI meal, a high-GI meal is characterized with hyperglycemia during the early postprandial stage (0-2h) and a compensatory hyperlipidemia associated with counter-regulatory hormone responses during late postprandial stage (4-6h). Over the past three decades, several human health disorders have been related to GI. The strongest relationship suggests that consuming low-GI foods prevents diabetic complications. Diabetic retinopathy (DR) is a complication of diabetes. In this aspect, GI appears to be useful as a practical guideline to help diabetic people choose foods. Abundant epidemiological evidence also indicates positive associations between GI and risk for type 2 diabetes, cardiovascular disease, and more recently, age-related macular degeneration (AMD) in people without diabetes. Although data from randomized controlled intervention trials are scanty, these observations are strongly supported by evolving molecular mechanisms which explain the pathogenesis of hyperglycemia. This wide range of evidence implies that dietary hyperglycemia is etiologically related to human aging and diseases, including DR and AMD. In this context, these diseases can be considered as metabolic retinal diseases. Molecular theories that explain hyperglycemic pathogenesis involve a mitochondria-associated pathway and four glycolysis-associated pathways, including advanced glycation end products formation, protein kinase C activation, polyol pathway, and hexosamine pathway. While the four glycolysis-associated pathways appear to be universal for both normoxic and hypoxic conditions, the mitochondria-associated mechanism appears to be most relevant to the hyperglycemic, normoxic pathogenesis. For diseases that affect tissues with highly active metabolism and that frequently face challenge from low oxygen tension, such as retina in which metabolism is determined by both glucose and oxygen homeostases, these theories appear to be insufficient. Several lines of evidence indicate that the retina is particularly vulnerable when hypoxia coincides with hyperglycemia. We propose a novel hyperglycemic, hypoxia-inducible factor (HIF) pathway, to complement the current theories regarding hyperglycemic pathogenesis. HIF is a transcription complex that responds to decrease oxygen in the cellular environment. In addition to playing a significant role in the regulation of glucose metabolism, under hyperglycemia HIF has been shown to increase the expression of HIF-inducible genes, such as vascular endothelial growth factor (VEGF) leading to angiogenesis. To this extent, we suggest that HIF can also be described as a hyperglycemia-inducible factor. In summary, while management of dietary GI appears to be an effective intervention for the prevention of metabolic diseases, specifically AMD and DR, more interventional data is needed to evaluate the efficacy of GI management. There is an urgent need to develop reliable biomarkers of exposure, surrogate endpoints, as well as susceptibility for GI. These insights would also be helpful in deciphering the detailed hyperglycemia-related biochemical mechanisms for the development of new therapeutic agents.
2010 Elsevier Ltd. All rights reserved.
Figures










References
-
- Abdallah W, Fawzi AA. Anti-VEGF therapy in proliferative diabetic retinopathy. Int Ophthalmol Clin. 2009;49:95–107. - PubMed
-
- Abordo EA, Thornalley PJ. Synthesis and secretion of tumour necrosis factor-alpha by human monocytic THP-1 cells and chemotaxis induced by human serum albumin derivatives modified with methylglyoxal and glucose-derived advanced glycation endproducts. Immunol Lett. 1997;58:139–147. - PubMed
-
- Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, Pasquale LR, Thieme H, Iwamoto MA, Park JE, Nguyen HV, Aiello LM, Ferrara N, King GL. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994;331:1480–1487. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
