

Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy
- PMID: 20871098
- PMCID: PMC3583518
- DOI: 10.1093/hmg/ddq419
Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy
Erratum in
- Hum Mol Genet. 2013 Apr 15;22(8):1697
Abstract
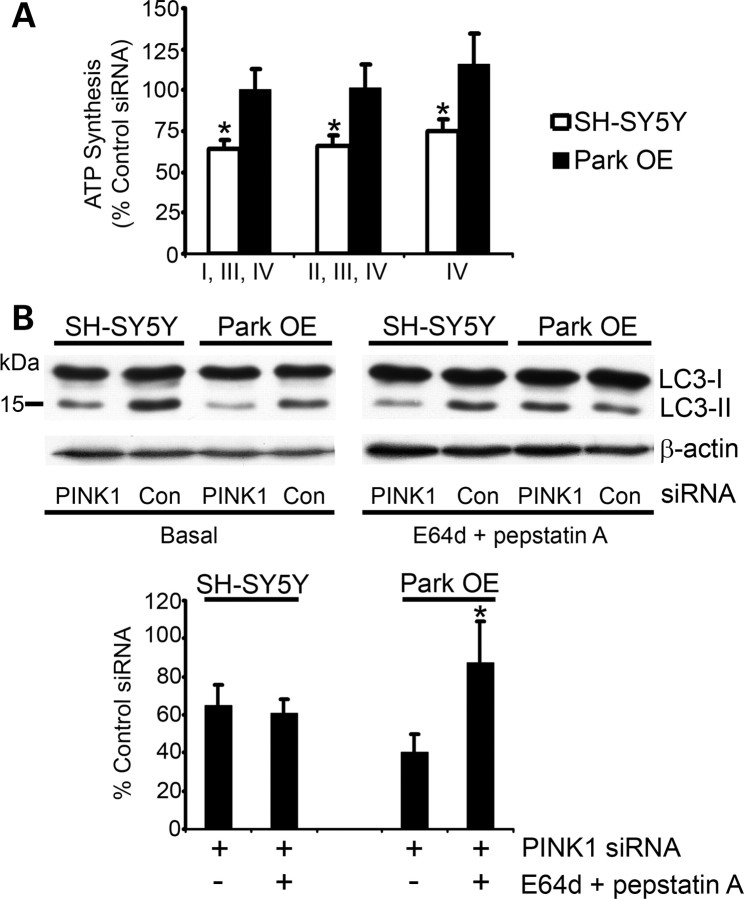
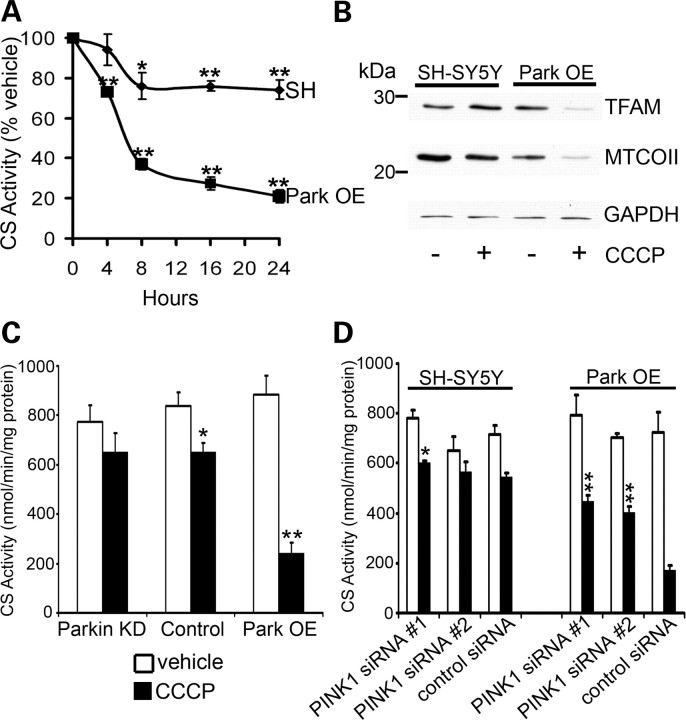
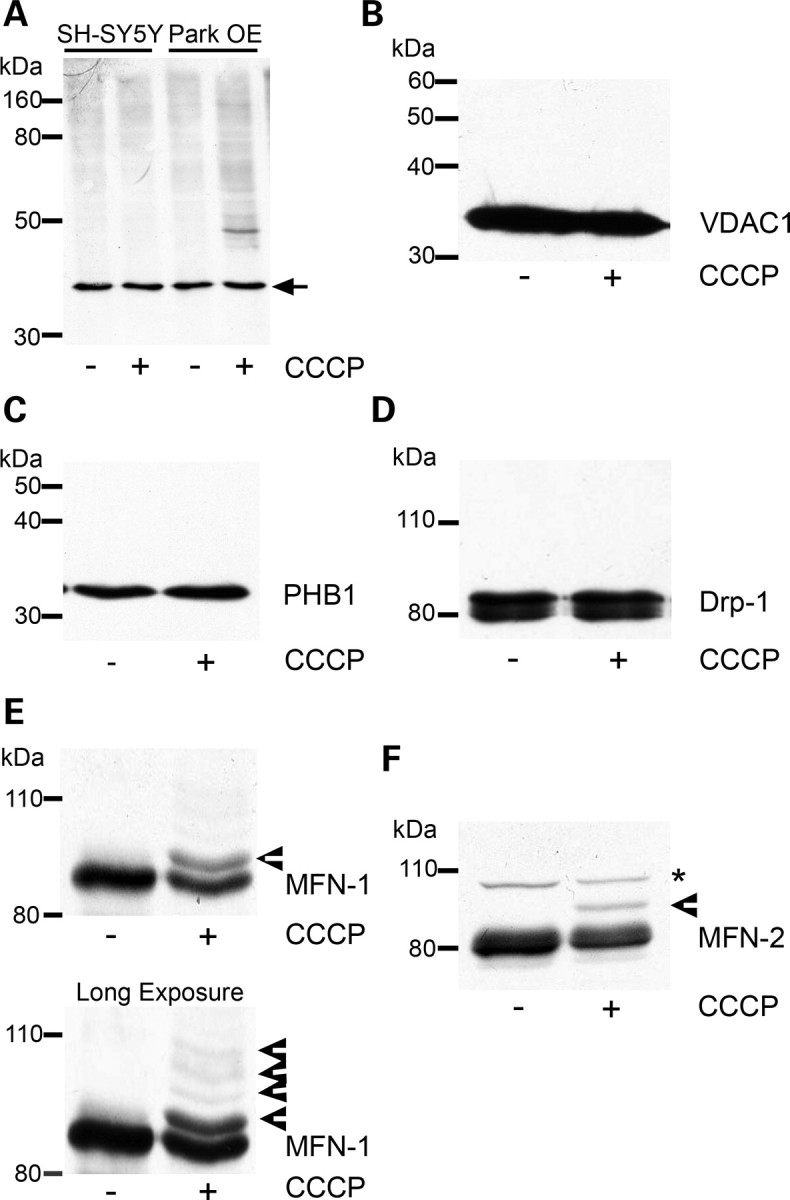
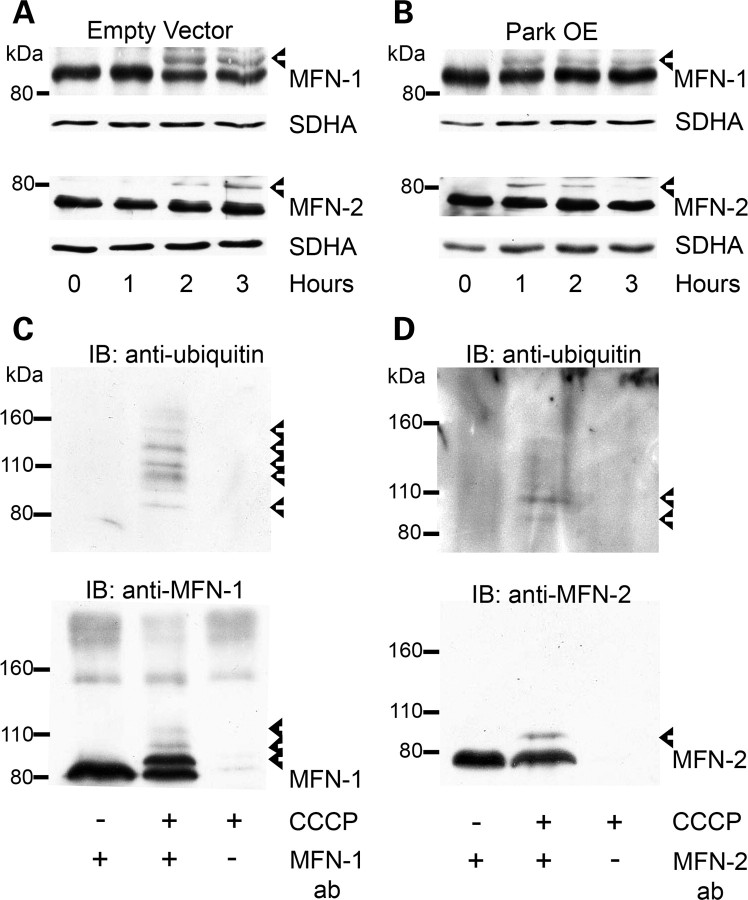
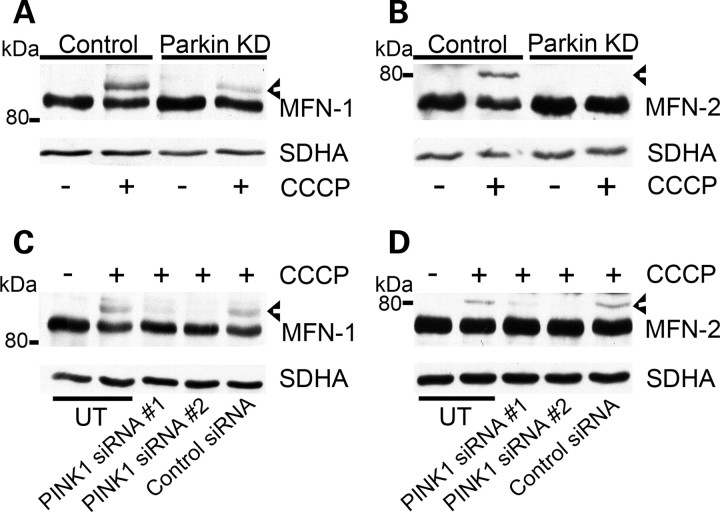
Mitochondrial dysfunction and perturbed degradation of proteins have been implicated in Parkinson's disease (PD) pathogenesis. Mutations in the Parkin and PINK1 genes are a cause of familial PD. PINK1 is a putative kinase associated with mitochondria, and loss of PINK1 expression leads to mitochondrial dysfunction, which increases with time. Parkin is suggested to be downstream of PINK1 and also mediates the removal of damaged mitochondria by macroautophagy (mitophagy). We investigated whether mitochondrial dysfunction in dopaminergic SH-SY5Y cells following decreased PINK1 expression by RNAi may in part be due to the inhibition of mitophagy. Reduced flux through the macroautophagy pathway was found to be coincident with the inhibition of ATP synthesis following 12 days of PINK1 silencing. Overexpression of parkin in these cells restored both autophagic flux and ATP synthesis. Overexpression and RNAi studies also indicated that PINK1 and parkin were required for mitophagy following CCCP-induced mitochondrial damage. The ubiquitination of several mitochondrial proteins, including mitofusin 1 and mitofusin 2, were detected within 3 h of CCCP treatment. These post-translational modifications were reduced following the silencing of parkin or PINK1. The ubiquitination of mitochondrial proteins appears to identify mitochondria for degradation and facilitate mitophagy. PINK1 and parkin are thus required for the removal of damaged mitochondria in dopaminergic cells, and inhibition of this pathway may lead to the accumulation of defective mitochondria which may contribute to PD pathogenesis.
Figures
References
-
- Schapira A.H., Tolosa E. Molecular and clinical prodrome of Parkinson disease: implications for treatment. Nat. Rev. Neurol. 2010;6:309–317. doi:10.1038/nrneurol.2010.52. - DOI - PubMed
-
- Cuervo A.M., Wong E.S., Martinez-Vicente M. Protein degradation, aggregation, and misfolding. Mov. Disord. 2010;25:S49–S54. doi:10.1002/mds.22718. - DOI - PubMed
-
- Schapira A.H., Cooper J.M., Dexter D., Jenner P., Clark J.B., Marsden C.D. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1989;1:1269. doi:10.1016/S0140-6736(89)92366-0. - DOI - PubMed
-
- Schapira A.H.V. Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol. 2008;7:97–109. doi:10.1016/S1474-4422(07)70327-7. - DOI - PubMed
-
- Betarbet R., Sherer T.B., MacKenzie G., Garcia-Osuna M., Panov A.V., Greenamyre J.T. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci. 2000;3:1301–1306. doi:10.1038/81834. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
