

Microfluidic platform for chemotaxis in gradients formed by CXCL12 source-sink cells
- PMID: 20871938
- PMCID: PMC4128891
- DOI: 10.1039/c0ib00041h
Microfluidic platform for chemotaxis in gradients formed by CXCL12 source-sink cells
Abstract
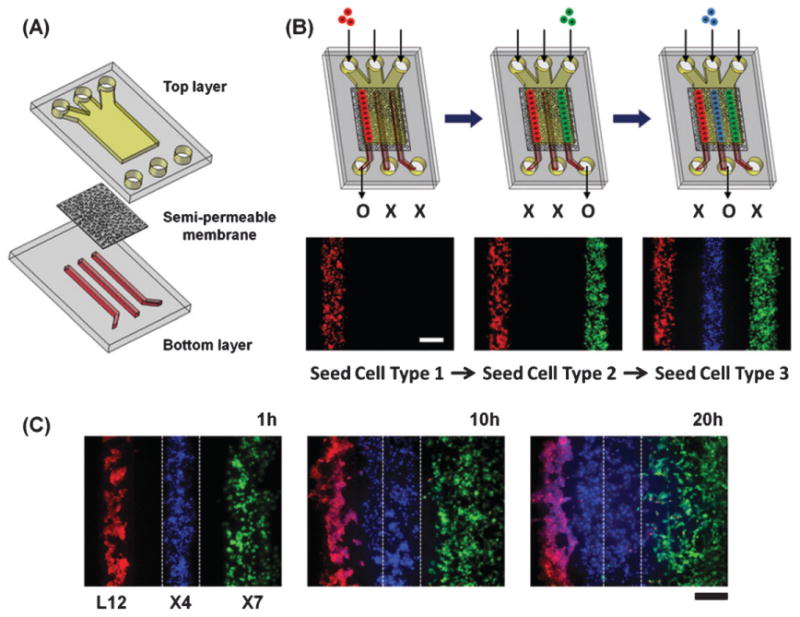
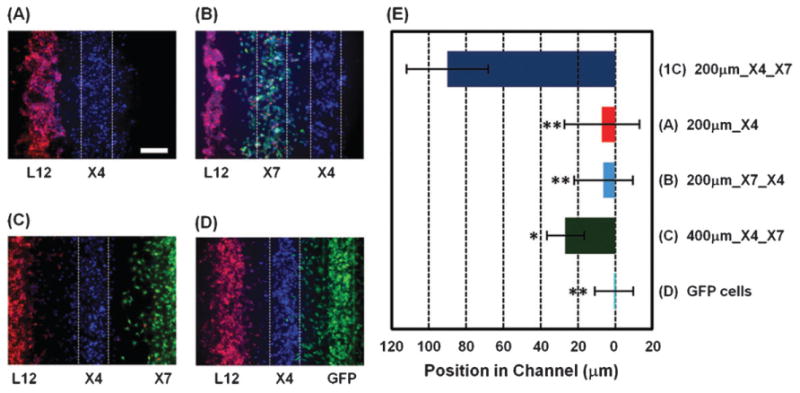
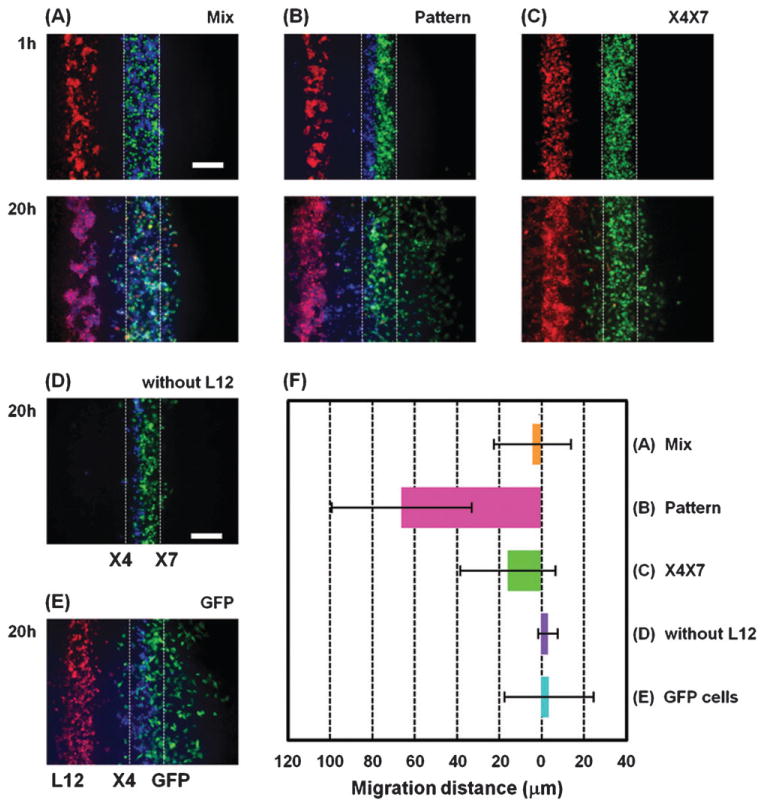
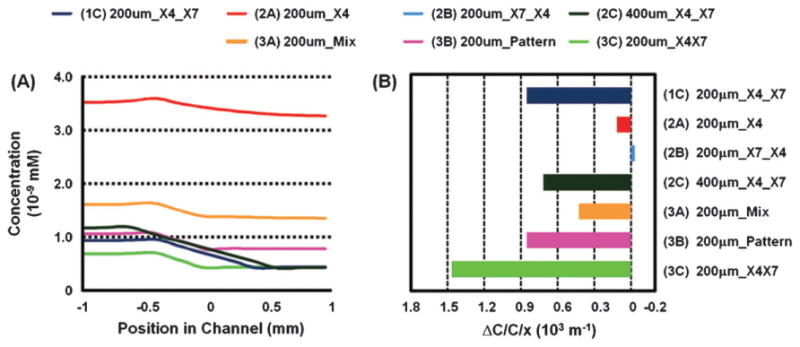
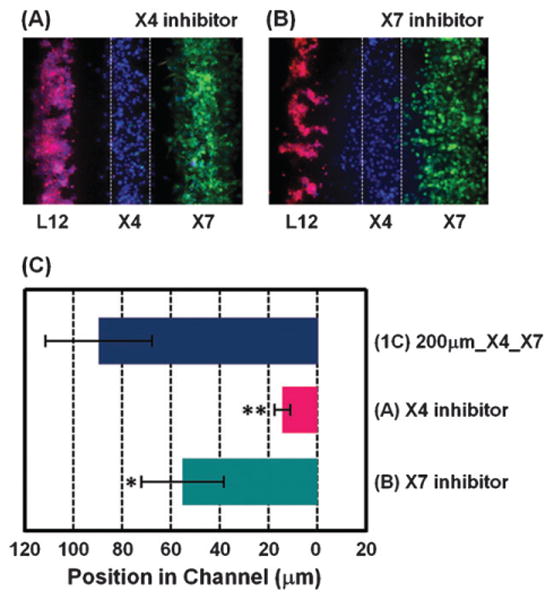
Chemokine CXCL12 promotes CXCR4-dependent chemotaxis of cancer cells to characteristic organs and tissues, leading to metastatic disease. This study was designed to investigate how cells expressing CXCR7 regulate chemotaxis of a separate population of CXCR4 cells under physiologic conditions in which cells are exposed to gradients of CXCL12. We recapitulated a cancer-stroma microenvironment by patterning CXCR4-expressing cancer cells in microchannels at spatially defined positions relative to CXCL12-producing cells and CXCR7-expressing cells. CXCR7 scavenges and degrades CXCL12, which has been proposed to facilitate CXCR4-dependent chemotaxis through a source-sink model. Using the microchannel device, we demonstrated that chemotaxis of CXCR4 cells depended critically on the presence and location of CXCR7 cells (sink) relative to chemokine secreting cells (source). Furthermore, inhibiting CXCR4 on migrating cells or CXCR7 on sink cells blocked CXCR4-dependent chemotaxis toward CXCL12, showing that the device can identify new therapeutic agents that block migration by targeting chemoattractant scavenging receptors. Our system enables efficient chemotaxis under much shallower yet more physiological chemoattractant gradients by generating an in vitro microenvironment where combinations of cellular products may be secreted along with formation of a chemoattractant gradient. In addition to elucidating mechanisms of CXCL-12 mediated chemotaxis, this simple and robust method can be broadly useful for engineering multiple microenvironments to investigate intercellular communication.
Figures
References
-
- Gurdon JB, Bourillot PY. Nature. 2001;413:797–803. - PubMed
-
- Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Nature. 2001;410:50–56. - PubMed
-
- Kunwar PS, Siekhaus DE, Lehmann R. Annu Rev Cell Dev Biol. 2006;22:237–265. - PubMed
-
- Friedl P, Weigelin B. Nat Immunol. 2008;9:960–969. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
